




















- DAZ.online
- News
- Apotheke
- Was bedeutet die neue EU-...
Herstellerpflichten
Was bedeutet die neue EU-Medizinprodukte-Verordnung für Apotheken? (Teil 2)
Köln - 27.05.2021, 11:45 Uhr

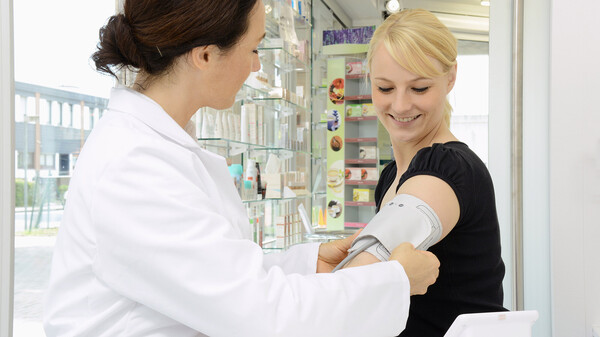
Der Vertrieb von Medizinprodukten, z. B. Halspastillen mit Hyaluronsäure, die das Apothekenlogo tragen, ist bei entsprechender vertraglicher Vereinbarung mit dem Hersteller weiterhin möglich, ohne dass die Apotheke zum Hersteller im Sinne der MDR wird. (Foto: IMAGO / Seelige)
Am gestrigen Mittwoch ist die Übergangsfrist für die neue EU-Medizinprodukteverordnung abgelaufen. Wir beleuchten, was die nun geltenden neuen Regelungen für Apotheken bedeuten. Während es im ersten Teil um Händlerpflichten ging, dreht sich der zweite und letzte Teil um Herstellerpflichten. Bei Ausführung welcher Tätigkeiten greifen diese? Außerdem wird erläutert, was bei einer Umkennzeichnung und Umverpackung von Medizinprodukten zu beachten ist.
Seit dem 26. Mai gilt EU-weit die neue Verordnung für Medizinprodukte (Medical Device Regulation – MDR). Zeitgleich ist das deutsche Medizinprodukterecht-Durchführungsgesetz (MPDG) in weiten Teilen in Kraft getreten. Wie das die Apotheken als „Händler“ von Medizinprodukten betrifft, konnten Sie bereits gestern auf DAZ.online lesen. Heute geht es um die „Herstellerpflichten“.
Mehr zum Thema

Ende der Übergangsfrist
Was bedeutet die neue EU-Medizinprodukte-Verordnung für Apotheken? (Teil 1)
Gemäß Artikel 16 Abs. 1 MDR kommen auf die Apotheke bei Ausführung folgender Tätigkeiten die Pflichten des Herstellers zu:
- Bereitstellung eines Produkts auf dem Markt unter dem eigenen Namen, dem eigenen eingetragenen Handelsnamen oder der eigenen eingetragenen Handelsmarke, außer in den Fällen, in denen eine Vereinbarung mit einem Hersteller geschlossen wurde, wonach der Hersteller als solcher auf der Kennzeichnung angegeben wird und für die Einhaltung der nach dieser Verordnung für die Hersteller geltenden Anforderungen verantwortlich ist. Der Vertrieb von Medizinprodukten, zum Beispiel Halspastillen mit Hyaluronsäure, unter Anbringung des Apothekenlogos, ist bei entsprechender vertraglicher Vereinbarung mit dem Hersteller weiterhin möglich, ohne dass die Apotheke zum Hersteller wird.
- Änderung der Zweckbestimmung eines bereits im Verkehr befindlichen oder in Betrieb genommenen Produkts;
- Änderung eines bereits im Verkehr befindlichen oder in Betrieb genommenen Produkts in einer Art und Weise, die Auswirkungen auf die Konformität des Produkts mit den geltenden Anforderungen haben könnte.
Von den oben genannten Änderungen hinsichtlich der Zweckbestimmung oder Änderungen, die Auswirkung auf die Konformität haben, sind Montagen und solche Anpassungen für den Patienten ohne Zweckänderungen des bereits in Verkehr gebrachten Medizinproduktes ausgenommen. Als Anwendungsfall wäre zum Beispiel an das Anpassen von Thrombosestützstrümpfen in der Apotheke zu denken.
Keine Herstellerpflichten bei den folgenden Tätigkeiten
Gemäß Artikel 16 Abs. 2 MDR gelten die folgenden Tätigkeiten nicht als Änderung des Produkts mit Einfluss auf die Konformität, dennoch bringen diese Tätigkeiten Händlerpflichten für die Apotheke mit sich:
- Bereitstellung, einschließlich Übersetzung, der vom Hersteller gemäß Anhang I Abschnitt 23 bereitzustellenden Informationen über ein bereits im Verkehr befindliches Produkt und weiterer Informationen, die für die Vermarktung des Produkts in dem jeweiligen Mitgliedstaat erforderlich sind. Demnach darf die Apotheke beispielsweise eine deutschsprachige Gebrauchsanweisung des Medizinprodukts zur Verfügung stellen, ohne dadurch zum Hersteller zu werden.
- Änderungen der äußeren Verpackung eines bereits im Verkehr befindlichen Produkts, einschließlich Änderung der Packungsgröße, falls das Umpacken erforderlich ist, um das Produkt in dem jeweiligen Mitgliedstaat zu vermarkten, und sofern dies unter Bedingungen geschieht, die gewährleisten, dass der Originalzustand des Produkts dadurch nicht beeinträchtigt werden kann. Bei Produkten, die steril in Verkehr gebracht werden, wäre der Originalzustand der Verpackung beeinträchtigt, wenn die zur Aufrechterhaltung der Sterilität notwendige Verpackung beim Umpacken geöffnet, beschädigt oder anderweitig beeinträchtigt wird. Auch durch das Auseinzeln oder Umverpacken von Medizinprodukten, wie etwa einzeln eingeschweißter Einmalspritzen, die aus der Aneinanderreihung abgetrennt werden müssen, gilt der Originalzustand als nicht beeinträchtigt. Bei importierten Produkten, die in anderen EU-Mitgliedstaaten nur in Großpackungen erhältlich sind, ist diese Tätigkeit des Apothekers für die Verkehrsfähigkeit aufgrund der in Deutschland zulässigen, kleineren Abpackungen erforderlich.
Beim Umpacken Hersteller und Behörde rechtzeitig in Kenntnis setzen
Die Apotheken trifft gemäß Artikel 16 Abs. 3 MDR eine Kennzeichnungspflicht bei den in Absatz 2 genannten Tätigkeiten: Daher müssen sie auf dem Produkt oder, falls dies nicht praktikabel ist, auf der Verpackung oder auf einem dem Produkt beiliegenden Dokument die Tätigkeit angeben, um die es sich handelt, sowie ihren Namen, ihren eingetragenen Handelsnamen oder ihre eingetragene Handelsmarke, ihre eingetragene Niederlassung und die Anschrift, unter der sie zu erreichen ist, sodass ihr tatsächlicher Standort ermittelt werden kann.
Voraussetzung für diese Tätigkeiten und die entsprechende Kennzeichnung ist die Etablierung und Einhaltung eines Qualitätsmanagementsystems (QMS), wie es in einer Apotheke auch für andere Aufgabenbereiche eingesetzt werden muss: Das QMS soll zum Beispiel sicherstellen, dass die Übersetzung der Informationen korrekt erfolgt und stets auf dem neuesten Stand ist. Zudem sollen die Tätigkeiten gemäß Absatz 2 mit Mitteln und unter Bedingungen durchgeführt werden, die gewährleisten, dass der Originalzustand des Produkts erhalten bleibt und die Verpackung des umgepackten Produkts nicht fehlerhaft, von schlechter Qualität oder unordentlich ist. Die Apotheke als Händler oder Importeur muss zudem über alle Korrekturmaßnahmen informiert werden, die der Hersteller in Bezug auf das betreffende Produkt als Reaktion auf Sicherheitsprobleme oder zur Herstellung der Konformität ergreift.
Umkennzeichnung und Umverpackung
Gemäß Artikel 16 Abs. 4 MDR müssen Apotheken mindestens 28 Tage bevor das umgekennzeichnete oder umgepackte Produkt auf dem Markt bereitgestellt wird, den Hersteller und die zuständige Behörde von ihrer Absicht in Kenntnis setzen. Darüber hinaus müssen Apotheken dem Hersteller und der zuständigen Behörde auf Verlangen eine Probe oder ein Modell des umgekennzeichneten oder umgepackten Produkts, einschließlich der übersetzten Kennzeichnung und der übersetzten Gebrauchsanweisung, zur Verfügung stellen. Dies bedeutet, dass die Apotheke beispielsweise sowohl über das Hinzufügen einer deutschsprachigen Gebrauchsanweisung als auch über das Umpacken von Medizinprodukten aus Großgebinden den Hersteller des betreffenden Medizinprodukts und die zuständige Behörde vorab informieren muss.
Ebenfalls erforderlich ist eine von einer Benannten Stelle (z. B. TÜV SÜD) ausgestellte Bescheinigung, in der gegenüber der zuständigen Behörde bescheinigt wird, dass das QMS der Apotheken den in Absatz 3 festgelegten Anforderungen entspricht.
Weitere Händlerpflichten der Apotheke
Zur Identifizierung innerhalb der Lieferkette sollen die Apotheken gemäß Artikel 25 Abs. 1 MDR mit den Herstellern oder ihren Bevollmächtigten zusammenarbeiten. Dadurch wird ein angemessenes Niveau der Rückverfolgbarkeit von Produkten angestrebt. In diesem Zusammenhang ist jeder Händler gemäß Absatz 2 verpflichtet, gegenüber der zuständigen Behörde Angaben zu machen über
- alle Wirtschaftsakteure, an die sie ein Produkt direkt abgegeben haben;
- alle Wirtschaftsakteure, von denen sie ein Produkt direkt bezogen haben;
- alle Gesundheitseinrichtungen oder Angehörigen der Gesundheitsberufe, an die sie ein Produkt direkt abgegeben haben.
Diese Verpflichtung gilt in einem Zeitraum von mindestens zehn Jahren, nachdem das letzte von der EU-Konformitätserklärung erfasste Produkt in Verkehr gebracht wurde. Da Apotheken Medizinprodukte vom Großhändler, der ein Wirtschaftsakteur ist, beziehen und die Produkte an Gesundheitseinrichtungen, wie Senioren- und Pflegeheime, Krankenhäuser oder Ärzte, direkt abgeben, kommt voraussichtlich ein erheblicher Verwaltungsaufwand auf sie zu, der sich nur mit entsprechenden Softwarelösungen bewältigen lassen wird.
Mehr zum Thema
Die zuständigen Behörden üben Marktüberwachungstätigkeiten aus und können gemäß Artikel 93 Abs. 3 MDR
- Wirtschaftsakteure verpflichten, die für die Zwecke der Durchführung der Tätigkeiten der Behörden erforderlichen Unterlagen und Informationen zur Verfügung zu stellen und, falls gerechtfertigt, die erforderlichen Produktstichproben kostenfrei bereitzustellen oder kostenfreien Zugang zu den Produkten zu ermöglichen, und
- angekündigte und erforderlichenfalls unangekündigte Kontrollen in den Räumlichkeiten der Wirtschaftsakteure sowie in den Räumlichkeiten von Zulieferern und/oder Unterauftragnehmern und, falls erforderlich, in den Einrichtungen beruflicher Anwender durchführen.
Insofern können auch Apotheken Adressaten solcher Maßnahmen und Kontrollen werden. Das Ergebnis des behördlichen Überwachungsberichts wird dem Wirtschaftsakteur gemäß Abs. 7 mitgeteilt und Gelegenheit zur Stellungnahme gegeben.
Was ist Apotheken nun zu raten?
Zusammenfassend ist den Apotheken zu raten, insbesondere das QMS der Apotheke mit den Vorgaben der MDR in Einklang zu bringen, um ab Ende Mai 2021 den neuen Händlerpflichten gerecht werden zu können. Bei der Qualitätssicherung können zur Unterstützung insbesondere zwei Leitlinien der BAK sowie die ergänzenden Kommentare und Flussdiagramme („Meldung von Vorkommnissen bei Medizinprodukten“) beziehungsweise Checklisten („Prüfkriterien für apothekenpflichtige Medizinprodukte“) herangezogen und für die eigene Apotheke adaptiert werden. Diese Leitlinien geben Empfehlungen zur Prüfung und Lagerung apothekenpflichtiger Medizinprodukte sowie zu Maßnahmen bei Risiken im Zusammenhang mit Medizinprodukten. Die jeweilige verwendete Apothekensoftware sollte dahingehend überprüft werden, ob sie eine lückenlose Dokumentation der eingegangenen Medizinprodukte im Sinne eines Warenwirtschaftssystems ermöglicht, um die Herkunft und Rückverfolgbarkeit über einen Zeitraum von zehn Jahren sicherzustellen.


0 Kommentare
Das Kommentieren ist aktuell nicht möglich.