














- DAZ.online
- News
- Politik
- Fortschritte bei der ...
Europäische Trilog-Verhandlungen
Fortschritte bei der Medizinprodukte-Verordnung
Berlin - 17.02.2016, 12:45 Uhr
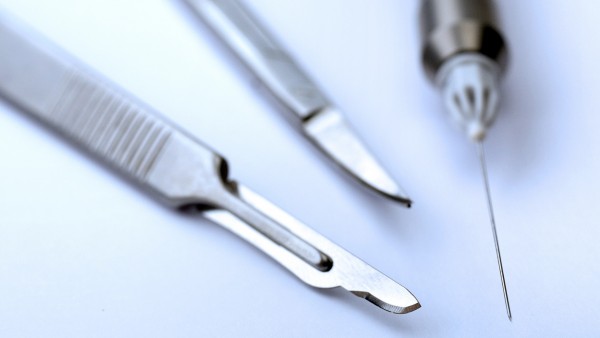
Auch über chirurgische Instrumente streitet man noch: In welche Risikogruppe fällt ein Skalpel? (Foto: euthymia/Fotolia)
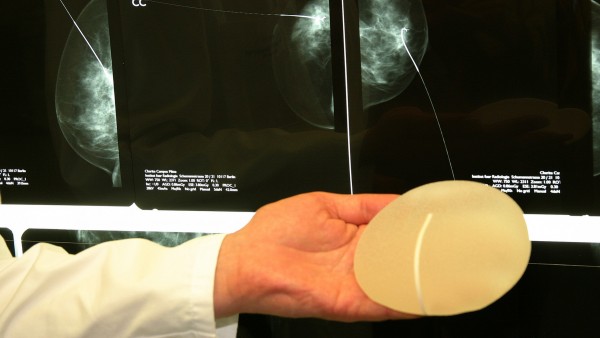
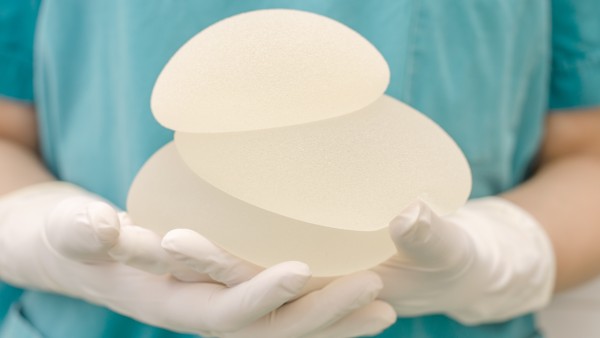
2011 und 2012 sorgte der Skandal um die mangelhaften Silikon-Brustimplantate der französischen Firma PIP für Schlagzeilen. Seitdem ist klar: Die europäischen Regelungen zu Medizinprodukten müssen überarbeitet werden. Nach zähen Verhandlungen ist die neue EU-Verordnung nun auf der Zielgeraden, sagt der EU-Parlamentarier Peter Liese (CDU).
Peter Liese von der Fraktion EVP-Christdemokraten im Europäischen Parlament ist zuversichtlich: Noch vor dem Sommer wird die neue EU-Medizinprodukteverordnung kommen. Ihr Weg war lang: 2012 hatte die Europäische Kommission ihren Verordnungsentwurf vorgelegt. Ein Jahr später hatte sich das EU-Parlament zu diesen Vorschlägen geäußert und eine Kompromissposition im Plenum beschlossen.
Dann waren die Mitgliedstaaten an der Reihe: Der Ministerrat einigte sich erst im Oktober 2015 auf eine gemeinsame Linie. Seitdem verhandeln Kommission, Parlament und Rat im sogenannten Trilog. Noch ist nicht in allen kontroversen Punkten eine Einigung erzielt – doch man kommt sich immer näher. Diese Woche Donnerstag wird erneut verhandelt. Wenn es sehr gut läuft, so Liese am Dienstag in Berlin, könne man sogar schon zu einer Einigung kommen.
Medizinproduktemarkt: groß und heterogen
Eine europäische Reglung zu Medizinprodukten ist angesichts des Binnenmarktes notwendig – der PIP-Skandal, der auch über Europa hinausging, zeigte deutlich, welche weitreichenden Folgen fehlerhafte Produkte haben können. Was sie zu einer Herausforderung macht, ist zum einen die schiere Masse der Medizinprodukte, etwa 500.000 gibt es auf dem europäischen Markt. Zudem fallen unter den Begriff des Medizinprodukts die unterschiedlichsten Gegenstände: Die Bandbreite reicht von wenig riskanten Lesebrillen und Mullbinden über schon kritischere Hörgeräte und Infusionspumpen bis hin zu hochriskanten Hüftprothesen, Stents und gar Herzschrittmachern. Eine eigene Verordnung bekommen In-vitro-Diagnostika, etwa Selbsttests zur Messung des Blutzuckerspiegels, HIV- und DNA-Tests. Hier soll die Regulierung aber anderen Medizinprodukten entsprechend erfolgen.
Gesundheitsgefährdende Risiken traten nicht nur bei PIP auf. „Auch bei Hüftimplantaten und Stents, die ins Gehirn eingepflanzt werden, gab es Probleme“, so Liese. „Daher müssen wir das System unbedingt stärken“. Auch bei In-vitro-Diagnostika machte man schon schlechte Erfahrungen. So seien jahrelang HIV-Tests aus Tschechien mit CE-Kennung im Umlauf gewesen, die wesentlich mehr falsch negative Ergebnisse anzeigten als andere Tests.
System der Benannten Stellen bleibt
Vorgesehen ist etwa eine strengere Kontrolle der sogenannten Benannten Stellen, die für die Prüfung und Zertifizierung der Medizinprodukte zuständig sind. In Deutschland sind dies etwa TÜV und DEKRA. Allerdings hat man sich entschieden, das System der Benannten Stellen beizubehalten und nicht – wie bei Arzneimitteln – staatliche Stellen mit den Aufgaben zu betrauen. Eine solche Übertragung wäre sehr aufwändig gewesen, erläuterte Liese, und hätte Skandale auch nicht sicher verhindern können. Schließlich gebe es auch bei Arzneimitteln immer wieder welche, erinnert sei etwa an Vioxx oder Mediator.
Neu ist auch, dass es künftig obligatorische Überprüfungen nach Inverkehrbringen des Produktes geben soll – genau solche fehlten bei den PIP-Implantaten. Weiterhin soll ein Implantatepass eingeführt werden, der für eine Rückverfolgbarkeit sorgt. Für Hochrisiko-Produkte soll es außerdem ein besonderes, anlassbezogenes Überwachungsverfahren geben.
Wo es noch hakt
Während sich die Trilog-Partner in diesen Punkten einig wurden, gibt es an anderen Stellen noch weiteren Diskussionsbedarf. Etwa bei der Frage der Haftpflichtversicherung für die Hersteller, die das Parlament möchte, der Rat aber bislang ablehnt. Das Argument der Gegner: Eine Haftpflichtversicherung hätte etwa im PIP-Fall auch nicht geholfen, da der Hersteller hier betrügerisch gehandelt hatte.
Diskussionen gab es auch noch bei den Klassifizierungen in die verschiedenen Risikoklassen sowie der Frage der klinischen Prüfungen. Das Parlament hätte hier gerne ein koordiniertes Verfahren der Mitgliedstaaten, wie es kürzlich für Arzneimittel-Studien beschlossen wurde. Das lehnt der Rat hingegen ab.
Doch Liese spricht hier von „Detailfragen“ – wie schnell sie wirklich zu lösen sind, wird sich möglicherweise schon diese Woche zeigen.


0 Kommentare
Das Kommentieren ist aktuell nicht möglich.