
Rolling-Review
Das Rolling-Review-Verfahren (fortlaufende oder gleitende Überprüfung) ist ein Regulierungsinstrument, das die EMA nutzen kann, um ein vielversprechendes Arzneimittel während eines Notfalls im Bereich der öffentlichen Gesundheit, wie beispielsweise der aktuellen Pandemie, zu bewerten. Normalerweise müssen alle zulassungsrelevanten Arzneimitteldaten zu Beginn des Evaluationsverfahrens eingereicht werden. Bei einer gleitenden Überprüfung werden die CHMP-Berichterstatter ernannt, während die Entwicklung noch im Gange ist, und die Agentur überprüft die Daten, sobald sie verfügbar sind.
















1 Kommentar
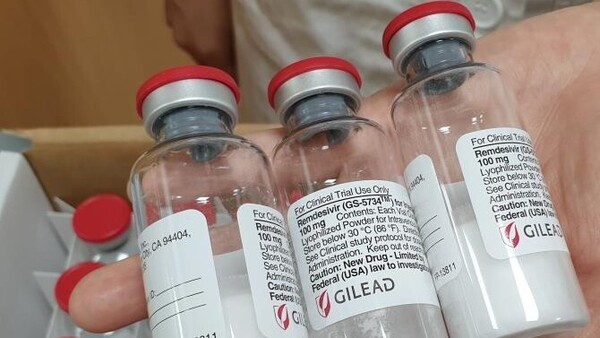
Remdesivir bisher fast ausschließlich in schweren Krankheitsverläufen eingesetzt
von Reinhold Schweitzer am 30.05.2020 um 14:56 Uhr
» Auf diesen Kommentar antworten | 0 Antworten
Das Kommentieren ist aktuell nicht möglich.