













- DAZ.online
- DAZ / AZ
- DAZ 22/2013
- Arzneimittel und CYP1A2
Klinische Pharmazie
Arzneimittel und CYP1A2
CYP1A-Subtypen
CYP1A2 macht zwar ca. 12% der hepatischen Cytochrom-P450-Enzyme aus, trägt aber zur Verstoffwechselung von nur ca. 3% der hepatisch eliminierten Pharmaka bei (s. 1. Beitrag dieser Serie). Exprimiert wird es im Intestinum und in der Leber.
Das strukturähnliche Isoenzym CYP1A1 wird überwiegend in der Lunge von Rauchern exprimiert und trägt z. B. zur Giftung von im Tabakrauch enthaltenen polyzyklischen aromatischen Kohlenwasserstoffen (PAK) wie dem Benz(a)pyren bei. CYP1A1 ist bei Nichtrauchern nicht nachweisbar.
Pharmakogenetik von CYP1A2
Es konnte eine große interindividuelle Variabilität hinsichtlich der Expression und Aktivität von CYP1A2 sowie der Elimination von Arzneistoffen, welche vornehmlich durch CYP1A2 metabolisiert werden, beobachtet werden. Verantwortlich hierfür sind wahrscheinlich genetische (z. B. SNPs), aber auch epigenetische Faktoren (z. B. DNA-Methylierung). CYP1A2 liegt polymorph vor, wobei es Varianten gibt mit herabgesetzter, normaler und erhöhter Aktivität sowie Varianten mit gesteigerter Induzierbarkeit. Die Auswirkungen der genetischen Varianten auf die Kinetik der Substrate sind jedoch weit geringer als bei CYP2D6. Darüber hinaus sind im klinischen Alltag die im Folgenden beschriebenen Arzneimittelwechselwirkungen relevanter.
Einige Studien deuten auf geschlechtsabhängige Unterschiede der CYP1A2-Aktivität hin [7, 10].
In eigener Sache: Zugang zu InteraktionsstudienEine genaue Bewertung von Arzneimittelrisiken und -interaktionen wird dadurch erschwert, dass die Hersteller häufig keine peer-reviewed, d. h. von anderen Forschern begutachtete, Publikation anstreben. Auf einige unserer Anfragen zu den selteneren Arzneimittelinteraktionen, die in Fachinformationen beschrieben worden sind, erhielten wir keine zufriedenstellenden Informationen zum Studienaufbau, zu den eingesetzten Dosierungen und den Ergebnissen. Ein Hersteller schrieb: "Studien mit zulassungsrelevanten Daten werden üblicherweise nicht immer in Fachjournalen publiziert, sondern liegen den Zulassungsbehörden in ausführlicher Form vor. […] Aus diesem Grund können wir Ihnen leider keine weiteren zitierbaren Quellen für diese Daten nennen." Die internen Studien wurden nicht herausgegeben. Somit kann zwar die Zulassungsbehörde die vorgelegten klinischen Daten kritisch bewerten; anderen Fachkreisen wird dies jedoch verwehrt. Dieser mangelhafte Informationsfluss führt dazu, dass wir manche Risiken nicht vollständig im klinischen Kontext bewerten oder gar konkrete Empfehlungen zur Dosisanpassung geben können. Zu befürchten ist vor allem eine falsch negative Pharmakovigilanz. Wissenschaftler, Apotheker und Ärzte kämpfen u. a. mit Petitionen wie der Berliner Erklärung 2012 (www.change.org > Suchen) oder www.alltrials.net gegen diese Intransparenz. |
Substrate von CYP1A2
CYP1A2 hat – wie CYP3A4 – zwar eine niedrige Affinität zu seinen Substraten, dafür aber eine hohe Kapazität und kann demnach nur bei sehr hohen Substratkonzentrationen gesättigt werden. Eine kompetitive Hemmung durch Substrate ist also selten, wurde aber am Beispiel des Coffeins in Einzelfällen beschrieben (s. u.).
CYP1A2 metabolisiert etliche wichtige endogene Verbindungen wie Steroide, Retinole, Melatonin und Arachidonsäure.
Pharmaka als Substrate
Als vorwiegend hepatisches Isoenzym ist CYP1A2 am oxidativen Metabolismus von Fluorchinolonen (Gyrasehemmern), Methylxanthinen, einigen Neuroleptika und Antidepressiva beteiligt. Da es nur über ein kleines aktives Zentrum verfügt, was durch mehrere sperrige aromatische Reste bedingt ist, handelt es sich bei seinen Substraten und Inhibitoren gewöhnlich um kleine lipophile und planare Moleküle.
Anhand der Metabolisierungsgeschwindigkeit des CYP1A2-Substrats Coffein kann die CYP1A2-Aktivität charakterisiert werden (Phänotypisierung). Eine präzise pharmakogenetische Vorhersage der Aktivität ist dagegen derzeit nicht möglich.
Es gibt nur wenige klinisch relevante Substrate von CYP1A2 (Tab. 1); darunter sind einige Arzneistoffe mit sehr geringer therapeutischer Breite wie Tizanidin oder Theophyllin, aber auch Arzneistoffe mit ausgeprägt großer therapeutischer Breite wie Agomelatin und Melatonin; wenn die Letzteren mit dem CYP1A2-Inhibitor Fluvoxamin kombiniert werden, steigt ihre Blutkonzentration zwar deutlich an (Tab. 2), unerwünschte Arzneimittelwirkungen (UAW) treten jedoch nicht auf.
Daneben ist CYP1A2 auch in die Giftung einiger Naturstoffe (Bildung toxischer Metaboliten) involviert. Ein Beispiel sind die nephrotoxischen und karzinogenen Aristolochiasäuren.
Tab. 1: Substrate, Inhibitoren und Induktoren von CYP1A2.
Tab. 2: Komedikation von CYP1A2-Inhibitor und -Substrat (Beispiele) und beobachtete Wirkung | ||
Inhibitor und Substrat mit Dosierungen [Literatur] |
Pharmakokinetische Auswirkung |
Klinische Auswirkung |
Ciprofloxacin 2 x 400 mg/d für 5 Tage und als Dauertherapie Clozapin 900 mg/d [11] |
Erhöhte Clozapin-Plasmakonzentration: 3 d nach Ciprofloxacin-Stopp und 1 d nach Clozapin-Stopp (wegen UAW): 890 ng/ml |
Rhabdomyolyse, Anstieg der Kreatinphosphokinase auf 195.000 U/L (normal < 200 U/L) |
Ciprofloxacin 500 mg/d und Tizanidin 4 mg/d [1] |
AUC von Tizanidin + 600% |
doppelt so starke Blutdrucksenkung |
|
Fluvoxamin und Agomelatin
[Fachinformationen Valdoxan®
,
keine Angaben zur Dosierung] |
Bioverfügbarkeit von Agomelatin 12 – 412-mal größer |
nicht beobachtet, da Agomelatin bereits in Normaldosis alle Rezeptoren sättigt |
Ciprofloxacin und Rasagilin [Fachinformationen Azilect® , keine Angaben zur Dosierung] |
AUC von Rasagilin + 83% |
nicht angegeben |
Umweltgifte als Substrate
Toxikologisch relevante Substrate von CYP1A1 und Induktoren beider CYP1A-Subtypen sind die polyzyklischen aromatischen Kohlenwasserstoffe (PAK) wie Benz(a)pyren [6]. Sie entstehen bei der unvollständigen Verbrennung (Pyrolyse) organischen Materials, also beispielsweise beim Tabakrauchen oder beim Grillen, wenn Fett auf die Kohle oder den Heizstab tropft. Einige PAK werden z. B. zu Epoxiden abgebaut und sind krebserzeugend. Heterozyklische aromatische Amine (HAA) entstehen durch Pyrolyse von Proteinen im Fleisch (z. B. Kruste). Sie sind krebserzeugend, denn sie werden durch CYP1A2 zu Hydroxylaminen abgebaut, die zu DNA-Schäden und damit zu Dysplasien und Tumoren führen.
Acrylamid entsteht durch die Reaktion von Aminosäuren und Stärke beim Bräunen von Backwaren oder Pommes frites. Sein kanzerogenes Potenzial ist unklar.
Nitrosamine bilden sich insbesondere beim Erhitzen von Nitrit-haltigen Lebensmitteln wie gepökelten Fleisch- und Fischprodukten, kommen aber auch im Tabakrauch vor. Sie sind stark krebserzeugend.
Alle genannten Prokanzerogene sind Substrate beider CYP1A-Subtypen:
- CYP1A1 giftet (= aktiviert) vor allem PAK in der Lunge,
- CYP1A2 giftet vor allem HAA in Leber und Darm.
Ein anderes Isoenzym, CYP2A6, ist maßgeblich an der Giftung von Nitrosaminen und dem Mykotoxin Aflatoxin B1 beteiligt. Das Letztere ist ein starkes Kanzerogen aus Aspergillus und kommt in kontaminiertem Getreide und Mehl vor. Bei Aflatoxin B1 bewirkt CYP1A2 keine Aktivierung, sondern eine Entgiftung.
Inhibitoren von CYP1A2
Typische kompetitive CYP1A2-Inhibitoren sind relativ kleine Moleküle, die häufig einen Methyl-, Chlor- oder Fluorsubstituenten besitzen (Tab. 1). Zu den starken Inhibitoren zählen das SSRI-Antidepressivum Fluvoxamin sowie die Gruppe der Fluorchinolon-Antibiotika. So können 750 mg Ciprofloxacin pro Tag die Bioverfügbarkeit von Coffein um 70% erhöhen. Der Ratschlag, unter Therapie mit Fluorchinolonen wegen potenzieller kardialer und zentralnervöser UAW weniger Coffein zu konsumieren, bleibt zwar aufgrund fehlender systematischer Untersuchungen umstritten, ein Hinweis auf die verstärkte Coffeinwirkung erscheint jedoch sinnvoll.
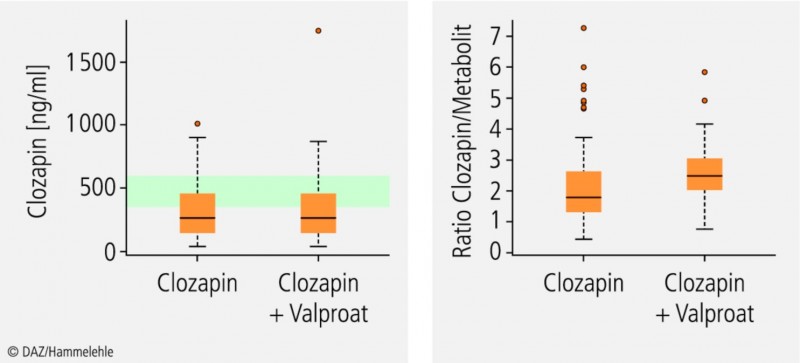
Coffein und Valproat sind schwache CYP1A2-Inhibitoren (Tab. 1). Valproat hemmt zwar bei Komedikation mit dem Neuroleptikum Clozapin dessen Metabolisierung, aber dies wirkt sich kaum auf die Plasmakonzentration von Clozapin aus und ist daher nicht klinisch relevant (Abb. 1).
Abb. 1:
Therapeutisches Drug Monitoring von Clozapin bei Monotherapie und bei Komedikation mit Valproat (ohne Valproat: n = 191; mit Valproat: n = 49); Plasmakonzentrationen (links) und metabolic ratio (rechts).
Durch die Komedikation verändert sich die Plasmakonzentration von Clozapin nicht, jedoch ist das Verhältnis (Ratio) von Clozapin zu seinem Metaboliten erhöht (Median ca. 2,7:1 statt ca. 1,8:1), was auf eine Enzymhemmung hindeutet. Der therapeutische Referenzbereich für Clozapin beträgt 350 bis 600 ng/ml (grüner Bereich); nach [13].
Auch schwache Inhibitoren können beim Vorliegen bestimmter genetischer Varianten oder bei Kombination (z. B. Valproat plus Coffein) die Plasmakonzentrationen von Arzneistoffen in klinisch relevantem Maße verändern; dies gilt insbesondere für Substrate mit geringer therapeutischer Breite wie Theophyllin.
Praxis-TippBei der Abgabe von starken CYP1A2-Inhibitoren (Fluorchinolone, Fluvoxamin, s. Tab. 1) an Patienten, die empfindlich auf Coffein reagieren oder größere Mengen Coffein konsumieren (Kaffee, Cola, Energy-Drinks, schwarze Schokolade), sollte der Apotheker darauf hinweisen, dass das Arzneimittel die Wirkung von Coffein verstärken kann. |
PAK als Induktoren von CYP1A2
Die Enzyme der Biotransformation bilden zusammen mit den Auswärtstransportern (z. B. P-Glykoprotein) eine kinetische Barriere, die den Organismus vor Fremdstoffen schützen sollen. Mehrere nukleäre Rezeptoren dienen unserem Körper einerseits als Sensoren, die Xenobiotika erkennen. Andererseits fördern diese Rezeptoren zugleich die Genexpression zur Synthese Arzneistoff-metabolisierender oder -transportierender Enzyme (s. 1. Beitrag dieser Serie). Dadurch kann die Enzym- bzw. Transportermenge innerhalb von Stunden bis Tagen dem aktuellen Bedarf angepasst werden.
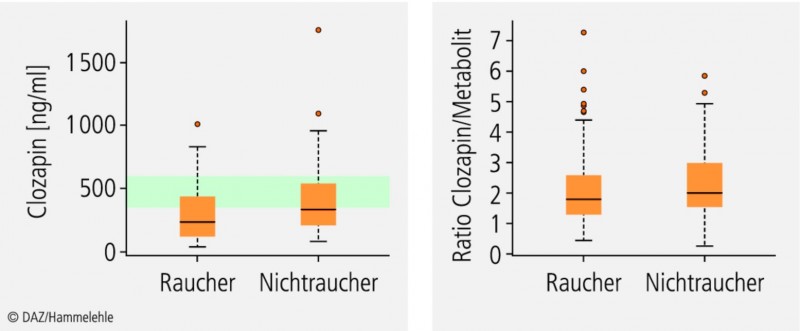
Abb. 2:
Therapeutisches Drug Monitoring von Clozapin bei Rauchern (n = 151) und Nichtrauchern (n = 93) ; Plasmakonzentrationen (links) und metabolic ratio (rechts).
Bei 67% der Raucher, aber nur 52% der Nichtraucher liegen die Plasmakonzentrationen von Clozapin unterhalb des therapeutischen Referenzbereiches (grün); der Unterschied zwischen beiden Gruppen ist signifikant (p < 0,01). Bei Plasmakonzentrationen oberhalb 1000 ng/ml Clozapin ist mit starken toxischen Wirkungen zu rechnen. – Das Verhältnis von Clozapin zu seinem Metaboliten (rechts) unterscheidet sich in beiden Gruppen hingegen nur geringfügig; nach [13].
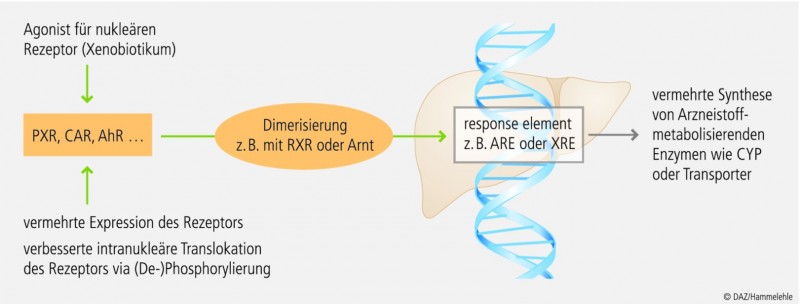
CYP1A2 kann – im Gegensatz zu Enzymen der CYP3A- oder CYP2C-Subfamilien – nicht über den Pregnan-X-Rezeptor (PXR) und nur moderat durch den konstitutiven Androstan-Rezeptor (CAR) induziert werden (Abb. 3). Die Gene der CYP1A-Subfamilie besitzen Bindungsstellen für den Arylhydrocarbonrezeptor (AhR). Wie der Name impliziert, sind insbesondere polyzyklische aromatische Kohlenwasserstoffe (PAK) Agonisten dieses Rezeptors. Dies erklärt, dass wir beim therapeutischen Drug Monitoring (TDM) von Rauchern signifikant niedrigere Plasmakonzentrationen von Clozapin gemessen haben (Abb. 2).
Abb. 3: Induktionsmechanismen
Nukleäre Rezeptoren wie PXR, CAR oder AhR können durch spezifische Agonisten wie Xenobiotika aktiviert werden, gelangen dann in den Zellkern und binden nach Dimerisierung an entsprechende response elements in der DNA. So verändern sie die Genexpression und Synthese von Cytochrom-P450-Isoenzymen und weiteren Proteinen; nach [8]. Die Genexpression von PXR und CAR kann z. B. durch Steroide erhöht werden.
Die Parameter der CYP1A2-Induktion durch PAK sind noch nicht im Detail bekannt. Das Ausmaß der Induktion ist anscheinend gewebespezifisch und auch von der Tabakart abhängig. Dabei korreliert die Anzahl der täglich gerauchten Zigaretten nur schwach mit der CYP1A2-Aktivität. Ausschlaggebend ist das Rauchen an sich und nicht seine Intensität. Bei Passivrauchern, die über längere Zeit täglich mehrere Stunden dem Zigarettenrauch exponiert sind, kann es ebenfalls zu einer CYP1A2-Induktion kommen.
Die CYP1A2-Induktion klingt nach dem Rauchstopp im Laufe von mehreren Monaten langsam ab. Dieser Umstand muss bei der Rauchentwöhnung von Patienten, die zugleich CYP1A2-Substrate wie Clozapin und weitere Neuroleptika oder auch andere Arzneistoffe (z. B. Riluzol) einnehmen, bedacht werden. Da das Nicotin bei den üblichen Dosierungen (wenige mg) keine klinisch relevante Enzyminduktion hervorruft [5, 6], muss bei der Substitutionstherapie mit Nicotin-Pflastern oder -Kaugummis die Dosis der CYP1A2-Substrate allmählich gesenkt werden. Das gilt auch für das Antiasthmatikum Theophyllin, dessen therapeutische Relevanz allerdings zunehmend schwindet.
Außer den PAK aktivieren auch die Inhaltsstoffe einiger Pflanzen wie Brokkoli und Rosenkohl die Expression von CYP1A2 (Tab. 1).
CYP1A2 spiegelt den Lebensstil wider
Die klinisch relevante Inhibition oder Induktion von CYP1A2 erfolgt weniger durch Medikamente als durch Genussmittel und -gifte: Bei Rauchern sind oft höhere Arzneimitteldosierungen zu wählen. Bei moderatem bis ausgeprägtem Coffeinkonsum (> 150 mg/d, > 2 Tassen Kaffee, s. Tab. 3) ist hingegen manchmal eine Dosisreduktion erforderlich, denn das Substrat Coffein kann zu einer leichten kompetitiven Hemmung von CYP1A2 führen.
Tab. 3: Gehalt an Coffein und anderen Methylxanthinen in Getränken, Genussmitteln und Arzneimitteln (Beispiele) | |
Produkt und Menge |
Methylxanthine |
1 Becher (ca. 200 ml) Kaffee (gemahlener Kaffee) |
70 – 120 mg |
1 Becher (ca. 200 ml) Instantkaffee |
40 – 65 mg |
1 Espressotasse (ca. 40 ml) Espresso |
30 – 50 mg |
1 Becher (ca. 200 ml) grüner oder schwarzer Tee |
30 – 120 mg |
1 Dose (330 ml) Cola oder Energy-Drink |
33 mg (Coca-Cola®) 82,5 mg (fritz-kola®) |
1 Portion (20 g) Schokolade mit 70% Kakaoanteil |
25 mg |
Standarddosis Theophyllin |
600 mg |
Weiterhin gilt, dass eine abrupte Veränderung im Konsum dieser Induktoren (Rauchen) bzw. Inhibitoren (z. B. Cola oder Kaffee) zu entsprechenden pharmakokinetischen Effekten führen kann. So kann ein durch eine Pneumonie bedingter Rauchstopp bei gleichzeitigem Beginn einer antibiotischen Therapie mit einem Fluorchinolon (CYP1A2-Inhibitor) die vor Therapiebeginn bestehende CYP1A2-Aktivität drastisch reduzieren.
Beratungshinweise
Der Apotheker sollte Kunden, die eine Rauchentwöhnung anstreben oder begonnen haben, nach ihrer eventuellen Dauermedikation fragen; relevant sind CYP1A2-Substrate wie Clozapin, Duloxetin, Theophyllin oder Tizanidin (Tab. 1). Da der durch den Tabakrauch bedingte CYP1A2-induzierende Effekt wegfällt, reduziert sich fortan die CYP1A2-Menge und verlangsamt sich der Metabolismus; deshalb muss eine früher erfolgte Dosiserniedrigung von CYP1A2-Substraten in Absprache mit dem Arzt wieder aufgehoben werden. Dies gilt auch bei der Nicotinsubstitution per Pflaster oder Kaugummi, da Nicotin selbst kein CYP1A2-Induktor ist.
Der klinische Fall
Das Therapeutische Drug Monitoring-Programm KONBEST (s. Infokasten im 2. Beitrag dieser Serie) bewertet ggf. auch den Einfluss von Arzneimittelinteraktionen auf gemessene unerwartete Plasmakonzentrationen. Im folgenden Fallbeispiel kann man die Auswirkung der Hemmung von CYP1A2 erkennen:
Ein 29-jähriger Psychotiker, 93 kg schwer, Raucher und Kaffeetrinker, wird mit 700 mg Clozapin und 10 mg Aripiprazol täglich behandelt (Tab. 4). Als UAW tritt im Laufe der Behandlung unnormales Schwitzen auf.
Tab. 4: Medikation, Abbauwege und mögliche Interaktionen im klinischen Fall. Modifiziert nach [14] | |||||
Wirkstoff |
CYP- |
||||
1A2 |
2C9 |
2C19 |
2D6 |
3A4 |
|
Clozapin |
X |
X |
X |
||
Aripiprazol |
X |
X |
|||
Coffein |
X |
||||
Rauchen |
X I |
||||
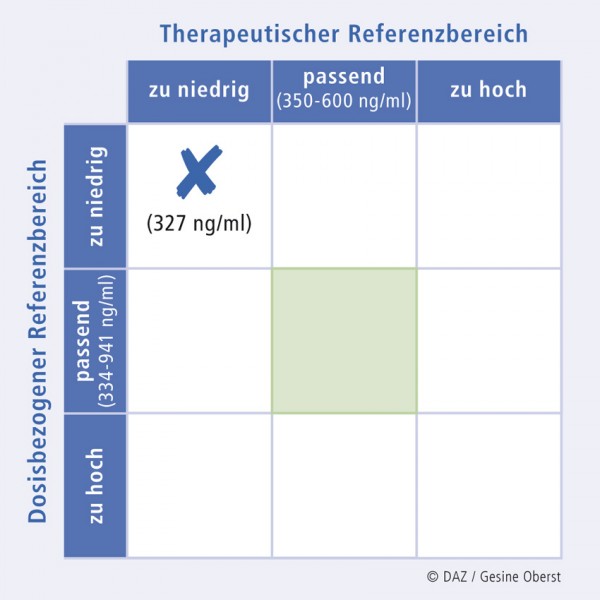
Eine Kontrolle der Plasmakonzentration von Clozapin ergibt einen Wert von 327 ng/ml, der sowohl unterhalb des angestrebten therapeutischen Referenzbereiches liegt (350 – 600 ng/ml; vgl. Abb. 1 und 2) als auch unterhalb des bei dieser Dosis erwarteten Wertes (334 – 941 ng/ml; Abb. 4).
Abb. 4:
Clozapin-Plasmakonzentration im klinischen Fall, dargestellt in der 9-Felder-Tafel; die Dosis beträgt 700 mg/d.
Die niedrige Plasmakonzentration lässt sich mit der Induktion von CYP1A2 durch Zigarettenrauch erklären, die einen schnelleren Abbau von Clozapin zur Folge hat. Der geringe Arzneistoffspiegel hat zu einem Nachlassen der anticholinergen Wirkung von Clozapin geführt, was sich bei dem Patienten durch stärkeres Schwitzen äußerte.
Literatur[1] Granfors MT, et al. Ciprofloxacin greatly increases concentrations and hypotensive effect of tizanidine by inhibiting its cytochrome P450 1A2-mediated presystemic metabolism. Clin Pharmacol Ther 2004;76(6):598 – 606.[2] Carrillo JA, et al. Effects of caffeine withdrawal from the diet on the metabolism of clozapine in schizophrenic patients. J Clin Psychopharmacol 1998;18(4):311 – 316.[3] Kirby BJ, et al. Complex drug interactions of HIV protease inhibitors 2: in vivo induction and in vitro to in vivo correlation of induction of cytochrome P450 1A2, 2B6, and 2C9 by ritonavir or nelfinavir. Drug Metab Dispos 2011; 39(12):2329 – 37. [4] Rote-Hand-Brief zu Arzneimittelinteraktionen zwischen Victrelis® (Boceprevir) und Ritonavir-geboosterten HIV-Proteaseinhibitoren. MSD, 23. 02. 2012. [5] Iba MM, et al. Dose-dependent up-regulation of rat pulmonary, renal, and hepatic cytochrome P-450 (CYP) 1A expression by nicotine feeding. Drug Metab Dispos 1999; 27(9):977 – 982.[6] Wei C, et al. Induction of CYP1A1 and CYP1A2 expressions by prototypic and atypical inducers in the human lung. Cancer Lett 2002;178(1):25 – 36.[7] Ou-Yang DS, et al. Phenotypic polymorphism and gender-related differences of CYP1A2 activity in a Chinese population. Br J Clin Pharmacol 2000;49(2):145-151.[8] Hukkanen J. Xenobiotic-metabolizing cytochrome P450 enzymes in human lung. Diss., Oulu 2001. [9] Sarich T, et al. The effect of omeprazole pretreatment on acetaminophen metabolism in rapid and slow metabolizers of S-mephenytoin. Clin Pharmacol Ther 1997;62(1):21-28.[10] Hakooz N, Hamdan I. Effects of dietary broccoli on human in vivo caffeine metabolism: a pilot study on a group of Jordanian volunteers. Curr Drug Metab 2007;8(1):9-15.[11] Brouwers EE, et al. Ciprofloxacin strongly inhibits clozapine metabolism: two case reports. Clin Drug Investig 2009;29(1):59 – 63. [12] Shin JG, et al. Effect of antipsychotic drugs on human liver cytochrome P-450 (CYP) isoforms in vitro: preferential inhibition of CYP2D6. Drug Metab Dispos 1999;27(9): 1078 – 84. [13] Hiemke C, et al. AGNP Consensus Guidelines for Therapeutic Drug Monitoring in Psychiatry: Update 2011. Pharmacopsychiatry 2011;44(6):195 – 235.[14] Flockhart, DA. Drug Interactions: Cytochrome P450 Drug Interaction Table. Indiana University School of Medicine, 2007. http://medicine.iupui.edu/clinpharm/ddis/table.aspx.
Autoren
Dr. med. Ruwen Böhm, Dr. rer. nat. Kirstin Reinecke,
Dr. rer. nat. Sierk Haenisch, Dr. Cornelia Börger,
Prof. Dr. med. Dr. rer. nat. Ingolf Cascorbi,
Prof. Dr. med. Thomas Herdegen
Institut für Experimentelle und Klinische Pharmakologie
Arnold-Heller-Str. 3, Haus 30, 24105 Kiel
E-Mail: ruwen.boehm@pharmakologie.uni-kiel.de
Prof. Dr. med. Dr. rer. nat. Ekkehard Haen, Abteilung Klinische Pharmakologie/Psychopharmakologie Psychiatrische Universitätsklinik Regensburg Bezirksklinikum Regensburg, Universitätsstraße 84, 93053 Regensburg
RückblickBisher sind in dieser DAZ-Serie folgende Beiträge erschienen: 1. Arzneimittelinteraktionen verstehen, vermitteln und vermeiden. DAZ 2012, Nr. 36, S. 64 – 74. 2. Interaktionen mit CYP3A4. DAZ 2012, Nr. 40, S. 58 – 62. 3. Arzneimittel und CYP2D6. DAZ 2012, Nr. 47, S. 60 – 66. |



0 Kommentare
Das Kommentieren ist aktuell nicht möglich.