













- DAZ.online
- DAZ / AZ
- DAZ 40/2012
- Interaktionen mit CYP3A4
Klinische Pharmazie
Interaktionen mit CYP3A4
Der Mensch besitzt vier CYP3A-Isoenzyme mit ähnlichen Eigenschaften – CYP3A4, CYP3A5, CYP3A7 und CYP3A43 – , von denen jedoch nur CYP3A4 und in geringerem Ausmaß CYP3A5 eine Rolle in der Pharmakokinetik spielen. CYP3A4 ist stark im Magen-Darm-Trakt und der Leber exprimiert und besitzt ein breites Substratspektrum. Es stellt daher eines der wichtigsten Abwehrsysteme gegen vor allem oral aufgenommene Xenobiotika dar.
Substrate von CYP3A4
Pharmaka, die überwiegend von CYP3A4 verstoffwechselt werden, sind in Tabelle 1 aufgeführt. Je höher die hepatische Clearance einer Substanz, desto niedriger ist nach oraler Applikation die in den Blutkreislauf aufgenommene Wirkstoffmenge (area under the curve, AUC), weil bereits ein großer Teil in der Leber und im Magen-Darm-Trakt metabolisiert und eliminiert wird (First-pass-Effekt). Bei Induktion von CYP3A4 wird die AUC solcher Pharmaka nochmals stark verkleinert (Tab. 2), während die Inhibition von CYP3A4 sich gegenteilig auswirkt. Bei parenteraler Applikation fällt der First-pass-Effekt weg und wirkt sich eine CYP3A4-Modulation weniger stark auf die AUC aus. Zu den endogenen Substraten von CYP3A4 gehören v. a. Steroide wie Sexualhormone und Vitamin D.
Tab. 1: Substrate, die überwiegend von CYP3A4 verstoffwechselt werden, und verwandte Substanzen, bei denen dies nicht (!) der Fall ist | |
|
Makrolidantibiotika
Clarithromycin
|
Erythromycin
|
|
Benzodiazepine
Alprazolam
Diazepam
Midazolam
|
Triazolam
nicht: Lorazepam,
Oxazepam |
|
Calciumkanalblocker
Amlodipin
Diltiazem
Felodipin
Lercanidipin
|
Nifedipin
Nisoldipin
Nitrendipin
Verapamil
|
|
Immunsuppressiva
Ciclosporin
Sirolimus
|
Tacrolimus
|
|
HIV-Protease-Inhibitoren
Indinavir
Ritonavir
|
Saquinavir
|
|
Statine
Atorvastatin
Lovastatin
|
Simvastatin
nicht: Pravastatin
|
|
Gerinnungshemmer
Apixaban
Phenprocoumon
Rivaroxaban
|
nicht: Dabigatran,
Heparin |
|
Diverse
Amiodaron
Aripiprazol
Buspiron
Carbamazepin
Chinidin
Chinin
Ethinylestradiol
|
Imatinib
Methadon
Sildenafil
Tamoxifen
Terfenadin
Trazodon
Vincristin
|
Tab. 2: Hepatische Clearance. Substrate von CYP3A4 mit unterschiedlicher hepatischer Clearance und der Einfluss einer CYP3A4-Induktion mit 600 mg/d Rifampicin über eine Woche auf die AUC (Daten aus [10]) | ||||
Substrat |
hepatische Clearance |
AUC (ng h/ml) |
Faktor |
|
ohne Rifampicin |
nach Induktion mit Rifampicin |
|||
Diazepam |
gering |
4430 |
1040 |
4,3 |
Midazolam |
mittel |
612 |
25 |
24,5 |
(S)-Verapamil |
maximal |
152 |
5 |
30,4 |
Inhibition von CYP3A4
Die Hemmung von Cytochrom-P450-Isoenzymen erfolgt, sobald der Hemmstoff im Darm- und Lebergewebe angeflutet ist, also in der Regel nach Sekunden (bei i. v. Applikation) oder wenigen Stunden (bei oraler Applikation). Die Dauer richtet sich nach der Art der Inhibition (Abb. 1). Bei inhibierenden Arzneistoffen mit langer Halbwertzeit, deren Wirkspiegel erst langsam aufgebaut werden, wie z. B. Amiodaron (HWZ 20 bis 107 Tage), können aber auch Wochen bis zur endgültigen Ausprägung der Arzneimittelinteraktion vergehen; und natürlich vergehen abermals Wochen, bis nach dem Absetzen des Medikaments keine Inhibition mehr vorhanden ist.
Kompetitive Hemmung – schnell und schwach ausgeprägt
Wichtig: Jedes Substrat von CYP3A4 belastet die Entgiftungskapazität und kann daher kompetitiv CYP3A4 hemmen. Oder anders gesagt: Ein CYP-Substrat ist ein kompetitiver CYP-Inhibitor. Eine kompetitive CYP3A4-Hemmung durch Nährstoffe und Medikamente ist aufgrund des großen Substratspektrums sehr häufig zu beobachten, jedoch wegen der oft nur schwachen Ausprägung meistens nicht klinisch relevant.
Cytochrom-P450-Enzyme – Evolution seit 3,5 Milliarden JahrenCytochrom-P450-Enzyme (CYP) sind nach ihrer Häm-Gruppe benannt, die Licht der Wellenlänge 450 nm absorbiert und deshalb rötlich erscheint (P = Pigment). CYP bilden eine große Superfamilie von Monooxygenasen und sind seit der Entstehung des Lebens vor ca. 3,5 Milliarden Jahren u. a. an der Biosynthese von Steroiden beteiligt. Abbau von XenobiotikaVor ca. 400 Millionen Jahren, zeitgleich mit der Ausbreitung von Pflanzenfressern (Herbivoren), kam es zum explosionsartigen Wachstum der CYP-Superfamilie in Tieren, denn sie bekamen eine neue Aufgabe: Da die Pflanzen sich mit der Bildung von sekundären Pflanzenstoffen wie Alkaloiden, Flavonoiden oder Terpenen vor Herbivoren zu schützen begannen, mussten die Tiere diese ihnen fremden (xenobiotischen) und potenziell schädlichen Stoffe abbauen bzw. entgiften können. Die Herbivoren standen unter einem Selektionsdruck, der zur Bildung zahlreicher CYP-Isoenzyme führte. Hinsichtlich ihrer CYP-Ausstattung weisen Tiere, die auf spezielle ökologische Nischen spezialisiert sind, die größten Besonderheiten und Unterschiede zu anderen Tieren auf. Individuelle UnterschiedeAuch innerhalb einer Spezies kann sich die CYP-Ausstattung individuell unterscheiden: So werden beim Menschen bezüglich CYP2D6 langsame, intermediäre, normale und schnelle Metabolisierer unterschieden. Die Häufigkeit der verschiedenen Metabolisierungstypen ist regional unterschiedlich (siehe nächster Beitrag dieser Serie). In der CYP3A-Subfamilie gibt es auch solche Varianten: CYP3A5 ist bei Europäern häufig inaktiv, bei Asiaten hingegen oft aktiv. So können Asiaten oft über CYP3A4 und CYP3A5 metabolisieren und benötigen z. B. deshalb höhere Dosen von Immunsuppressiva. Veränderung der CYP-AktivitätDie pflanzlichen "Gifte" sind nicht nur Substrate für CYP, sondern können auch CYP induzieren oder inhibieren. Beispiele sind das Flavonoid Naringin und das Cumarin Bergamottin aus Schale und Kernen der Grapefruit (Citrus paradisi), die beide CYP3A4 und andere CYP inhibieren, oder Hyperforin aus dem Johanniskraut (Hypericum perforatum), welches CYP3A4 und andere CYP induziert. Die Liste dieser CYP-Funktion-verändernden Substanzen ist lang und reicht von "normalen" Nahrungsmitteln wie Lauchgewächsen (Knoblauch), Brokkoli, Pfefferminzöl und Zitrusfrüchten über exotischere Nahrungsmittel wie Ingwer oder Lakritze und Genussmittel/-gifte wie Alkohol, Nicotin und Cannabidiol zu Nutrizeutika und Phytopharmaka wie Johanniskraut, Kanadischer Gelbwurz, Wu Wei Zi (Schisandra chinensis), Ginseng und Baldrian. Auch aus anderen, nicht-pflanzlichen Organismen gewonnene Pharmaka (z. B. Antibiotika bakteriellen Ursprungs wie Rifampicin) und durch unvollständige Verbrennung entstandene Gifte (z. B. Tabakrauch, angekohltes Fleisch) verändern die CYP-Aktivität. Nicht alle Induktionen und Inhibitionen von CYP sind klinisch relevant: Für die meisten Substanzen wurden zwar in vitro Inhibitionsphänomene nachgewiesen, diese werden aber häufig in vivo von anderen Faktoren (z. B. gleichzeitige Induktion oder nur geringe Bedeutung des inhibierten CYP für den Metabolismus) überlagert. Bei anderen Substanzen müssen größere Mengen eingenommen werden, um eine klinisch relevante Veränderung der CYP-Aktivität zu erreichen (z. B. 200 ml Grapefruitsaft, 600 mg Johanniskrautextrakt, 300 g gegrilltes Fleisch pro Tag über mehrere Tage, zehn Zigaretten pro Tag). Die ursprüngliche Aufgabe der CYP, Steroide für Zellmembranen und für das Hormonsystem zu synthetisieren, zeigt sich auch immer wieder in der Praxis: Azolantimykotika wie Ketoconazol, die in Pilzen die CYP51-vermittelte Synthese des Zellmembranbestandteils Ergosterol hemmen, inhibieren auch Isoenzyme der CYP1-, CYP2- und CYP3-Familien, die vor allem Arzneistoffe metabolisieren. Andererseits interagieren Hormonpräparate besonders häufig mit Substanzen, die die CYP-Aktivität verändern (z. B. Interaktion der "Pille" mit Carbamazepin). |
Irreversible Hemmung – langsam, stark ausgeprägt und langandauernd
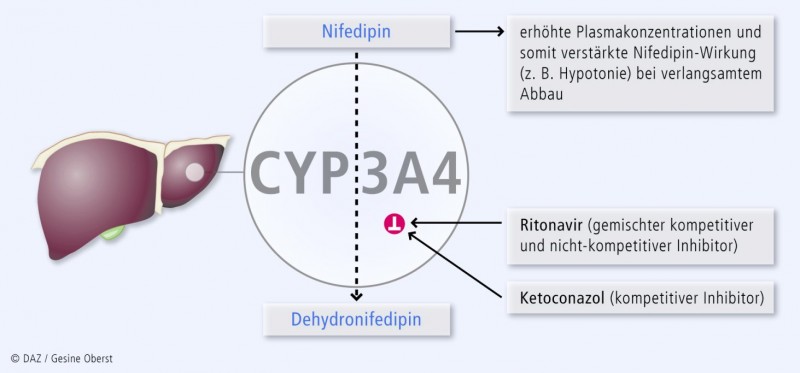
Über die kompetitive Hemmung hinaus kann CYP3A4 irreversibel gehemmt werden; man spricht auch von nicht-kompetitiver Hemmung oder Suizidhemmung. Hierbei entstehen in der Regel Metaboliten, die kovalent an das Cytochrom-Apoprotein oder das Häm binden. Da dieser Hemmmechanismus eine vorherige Umsetzung der Muttersubstanz zum hemmenden Metabolisierungsprodukt erfordert, wird er auf Englisch auch als "time-dependent inhibition" oder "mechanism-based inhibition" bezeichnet. Diese Inhibitoren – obgleich zahlenmäßig weitaus seltener als kompetitive Hemmstoffe – haben eine wesentlich größere klinische Relevanz. Der irreversible Inhibitionsmechanismus hält unabhängig von der Plasmakonzentration des Inhibitors an, bis die irreversibel gehemmten CYP neu synthetisiert worden sind (HWZ s. u.). Eine zeitlich versetzte Einnahme von Substrat und Inhibitor (z. B. Nifedipin und Grapefruitsaft) ist somit keine Lösung des Problems.
Abbildung 1 zeigt diesen Mechanismus beispielhaft. In Tabelle 3 sind Inhibitoren von CYP3A4 aufgeführt. Die Stärke der Enzymhemmung ist semiquantifiziert in drei Stufen eingeteilt.
Tab. 3: Klinisch relevante Inhibitoren von CYP3A4* (Stärke der Inhibition: + bis +++). Die Inhibition ist in den meisten Fällen irreversibel, oder sie hat, wie bei Ketoconazol, eine lange Halbwertzeit. | |
|
HIV-Protease-Inhibitoren
Indinavir +++
Nelfinavir +++
|
Ritonavir +++
|
|
Makrolidantibiotika
Clarithromycin +++
Erythromycin ++
|
nicht: Azithromycin
|
|
Azolantimykotika
Ketoconazol +++
Fluconazol ++
|
Itraconazol ++
Voriconazol +
|
|
Diverse
Aprepitant ++
Cimetidin ++
Verapamil ++ (vgl. Tab. 2)
Naringin, Bergamottin ++
(in Citrusfrüchten,
v. a. Grapefruit und Bitterorange) Kanadische Gelbwurz +
|
Amiodaron +
Ciclosporin +
Diltiazem +
Fluvoxamin +
Norfluoxetin +
(aus Fluoxetin)
nicht: Baldrian, Mariendistel, Ginkgo
|
* Der genaue Einfluss der Inhibition auf die Therapie ist immer von der Konzentration des Inhibitors, des Substrates und der Eliminationswege des Substrates abhängig.
Besondere Aspekte der Inhibition von CYP3A
Intrauterin wird auch das Isoenzym CYP3A7 exprimiert, welches die fötale Entwicklung mitsteuert. Die gleichzeitige Gabe von v. a. CYP3A4-Inhibitoren, die auch stark CYP3A7 hemmen, wie den HIV-Proteaseinhibitoren Nelfinavir und Ritonavir kann somit den Fötus schädigen.
Die Inhibition von CYP3A4 ist nicht immer unerwünscht: Mithilfe von Ritonavir kann z. B. die Konzentration anderer HIV-Protease-Inhibitoren in Lymphozyten erhöht werden, was die HIV-Behandlung verbessert und mittlerweile fester Bestandteil der HIV-Therapie ist (sog. Ritonavir-Booster). Im Einzelfall kann mit den CYP3A4-Inhibitoren Verapamil oder Ketoconazol die benötigte Dosis des teuren Immunsuppressivums Ciclosporin A gesenkt werden.
Induktion von CYP3A4
Induktionseffekte beruhen auf einer erhöhten Menge von CYP-Isoenzymen in Leber und Darm. Induktions- und Deinduktionseffekte (Absetzen des Induktors) treten daher zeitverzögert auf: Die Synthesedauer von CYP wird mit ca. 24 bis 48 Stunden, die Abbau-HWZ mit ca. 30 Stunden angegeben. Dementsprechend sind erste Induktionseffekte bereits nach wenigen Stunden nachweisbar, und eine maximale Induktion ist nach ca. 5 Tagen erreicht. Bei Deinduktion muss ca. 6 bis 7 Tage gewartet werden, bis die basale CYP-Menge wieder erreicht ist.
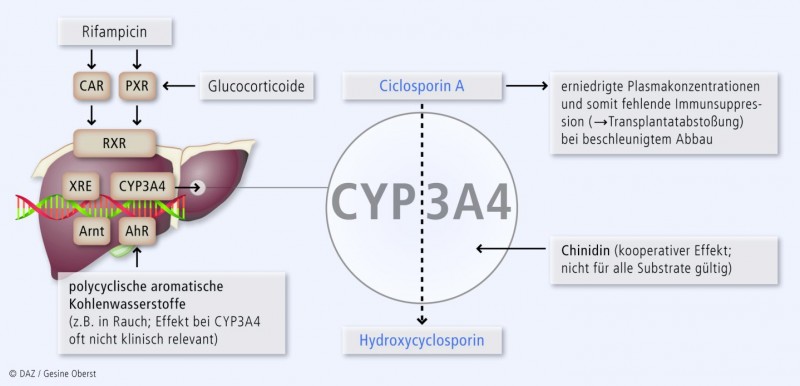
Induktionsmechanismus für ein synergistisches Entgiftungssystem mit P-gp
Die Gene für CYP3A-Isoenzyme und den Arzneimittel-eliminierenden Effluxtransporter P-Glykoprotein (P-gp, Genname: ABCB1) liegen dicht beieinander. Ihre Expression wird jeweils durch xenobiotic response elements (XRE) in ihrem Promotor kontrolliert. Diese regulatorischen XRE werden durch Transkriptionsfaktor-Komplexe aktiviert, die – in Analogie zur Wirkung von Steroidhormonrezeptoren und ihren Liganden – aus nukleären Rezeptoren bestehen wie dem Pregnan-X-Rezeptor (PXR, syn. Steroid-X-Rezeptor, SXR; Genname: NR112) und dem konstitutiven Androstanrezeptor (CAR, Genname: NR1I3). Damit diese Rezeptoren an die DNA binden, müssen sie ihrerseits von Liganden aktiviert werden wie den in Tab. 4 aufgeführten Xenobiotika. Als Konsequenz der identischen Regulation induzieren diese PXR-Agonisten nicht nur die CYP3A-Isoenzyme, sondern auch P-gp und bilden so ein synergistisches Entgiftungssystem (Abb. 2).
Tab. 4: Induktoren von CYP3A4 und P-gp* (Stärke der Induktion: + bis +++) | |
|
Carbamazepin ++
Oxcarbazepin +
Dexamethason ++
nicht: Hydrocortison, Prednisolon
Phenobarbital +++
Phenytoin +++
Rifampicin +++
|
Efavirenz +
Hyperforin ++
(in Johanniskraut) nicht: Crataegus, Echninacea, Cimicifuga, Ginseng
|
* Der genaue Einfluss der Induktion auf die Therapie ist immer von der Konzentration des Induktors, des Substrates und der Eliminationswege des Substrates abhängig.
Praxis-TippZu den klinisch relevanten, starken Inhibitoren von CYP3A4 zählen v. a. Antiinfektiva (Virostatika, Makrolidantibiotika, Azolantimykotika) und Verapamil (Tab. 3). Die Dauer der Inhibition richtet sich nach der Halbwertzeit des Inhibitors und der Dauer für die Neusynthese des irreversibel gehemmten CYP-Isoenzyms. |
Induktion durch Johanniskraut
Neueren Studien zufolge ist der Einfluss von Johanniskraut umstritten. Zwar kann eindeutig eine größere Clearance zahlreicher Pharmaka unter Therapie mit Johanniskrautextrakten beschrieben werden, deren Auswirkungen auf die pharmakodynamische Wirkung dieser Pharmaka sind jedoch geringer als bisher postuliert. Weitere Details folgen in einem späteren Beitrag dieser Serie.
Praxis-TippZu den Induktoren von CYP3A4 zählen vor allem Antikonvulsiva wie Carbamazepin, Phenytoin und Phenobarbital (Tab. 4). Die Induktion und Deinduktion benötigen jeweils einige Tage bis zur vollen Ausprägung. |
Induktion und Steroidstoffwechsel
Eine langanhaltende Induktion von CYP3A via PXR kann zu weiteren UAW führen: Auch Steroide wie Sexualhormone oder Vitamin D werden über CYP3A verstoffwechselt. So ist z. B. unter Phenobarbital- oder Phenytointherapie die Elimination von Vitamin D beschleunigt. Die Konsequenz können Rachitis oder Osteomalazie sein. Des Weiteren ist die Wirkung der oralen Kontrazeptiva beeinträchtigt.
Kombinierte Induktion und Inhibition
Einige Substanzen, die die CYP-Aktivität verändern, sind kombinierte Inhibitoren und Induktoren (Substrat = Inhibitor = Induktor). Klinisch überwiegt bei Dauertherapie immer einer der beiden Effekte. Beispiele beim CYP3A4 sind Rifampicin und Ritonavir: Beide sind sowohl Induktor als auch Inhibitor von CYP3A4. Bei Rifampicin ist die inhibitorische Wirkkomponente nur zu Therapiebeginn kurzzeitig nachweisbar, bevor die sehr starke Induktion überwiegt. Bei Ritonavir ist die inhibitorische Wirkung weitaus größer als die eher geringe Enzyminduktion, sodass hier immer die Inhibition überwiegt.
Klinische Fälle
Programme zum Therapeutischen Drug Monitoring (TDM, s. Infobox) bewerten gegebenenfalls auch den Einfluss von Arzneimittelinteraktionen auf gemessene unerwartete Plasmakonzentrationen. Die folgenden Fälle 1 und 2 zeigen, welche Auswirkung eine Inhibition bzw. Induktion von CYP3A4 haben kann.
Therapeutisches Drug Monitoring mithilfe von KONBESTKONBEST (abgeleitet von "Konzentrationsbestimmung") ist eine Internetplattform zur Erfassung, Organisation und klinisch-pharmakologischen Befundung von Wirkstoffkonzentrationsbestimmungen. Der Einsender der jeweiligen Blutprobe bzw. der bereits ermittelten Konzentration erhält eine pharmakologische Bewertung unter Berücksichtigung der Patientendaten (Alter, Geschlecht, Gewicht), der Diagnose, des Therapieverlaufs, der UAW und der Komedikation. Hierdurch können individuelle Faktoren des Patienten wie seine Compliance und Veränderungen des Arzneimittelstoffwechsels, die auf Interaktionen mit Arznei-, Genuss- und Nahrungsmitteln sowie auf genetischen Besonderheiten (langsame bzw. schnelle Metabolisierer) beruhen, erkannt werden. Ein großes Team freiwilliger Helfer (z. B. Assistenzärzte) wirkt an diesen Befundungen mit und trägt so zur Optimierung der Pharmakotherapie bei. Befunder nutzen KONBEST zur eigenen supervidierten Fort- und Weiterbildung. Werkzeuge für die Befundung sind u. a. der Dosis-bezogene Referenzbereich, der therapeutische Referenzbereich und die sich daraus ergebende 9-Felder-Tafel ([6], Abb. 3 und 4) sowie eine Tabelle mit den Stoffwechselwegen der gesamten Medikation (Tab. 5 und 6). Eventuell vorhandene Vorbefunde und die metabolic ratio können ebenfalls in die Bewertung einfließen [7]. |
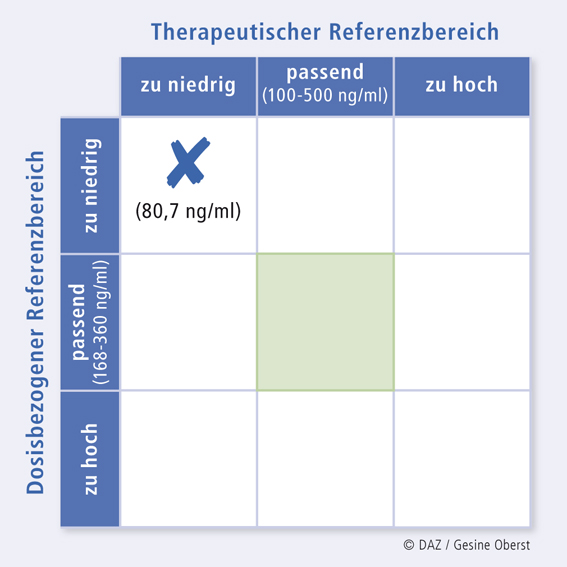
Fall 1
Ein 39-jähriger, 110 kg schwerer Patient mit bipolarer Persönlichkeitsstörung, Raucher, erhält 600 mg Quetiapin täglich zur Stimmungsstabilisierung. Das Ansprechen auf die Pharmakotherapie ist jedoch mäßig. Im Blutplasma wurde eine Quetiapinkonzentration von 80,7 ng/ml gemessen, was unterhalb des therapeutischen Referenzbereiches liegt (Abb. 3). Die gemessene Plasmakonzentration liegt auch unterhalb des in Relation zur Dosis erwarteten Fensters (sog. Dosis-bezogener Referenzbereich, Abb. 3).
Außer Quetiapin erhält der Patient noch zwei weitere Medikamente (Tab. 5). Quetiapin hat mit Clonazepam und Carbamazepin gemeinsame Stoffwechselwege (CYP2D6, CYP3A4/5), die durch Carbamazepin und Zigarettenrauch induziert werden.
|
Tab. 5: Aktuelle Medikation, Abbauwege und mögliche Interaktionen zu Fall 1 Modifiziert nach [4] sowie Fachinformationen | |||||
Wirkstoff |
CYP- |
||||
1A2 |
2C9 |
2C19 |
2D6 |
3A4 |
|
Quetiapin |
X |
X |
|||
Carbamazepin |
X I |
||||
Clonazepam |
X |
||||
Zigarettenrauch |
X I |
||||
Wie ist also diese geringe Plasmakonzentration zu erklären? Neben der Noncompliance, welche grundsätzlich eine häufige Ursache ist, ist hier eine Enzyminduktion von CYP3A4 durch Carbamazepin sehr wahrscheinlich.
Bei eventuellen Dosisänderungen ist zu beachten, dass der Patient unter der angegebenen Komedikation eine flachere Dosis-Konzentrations-Beziehung zeigt. Anstieg oder Abfall der Plasmakonzentration bei Änderungen der Dosis werden also schwächer ausfallen.
Zu erwarten ist auch eine auf die Dosis bezogene erniedrigte Plasmakonzentration von Clonazepam mit entsprechender Wirkungsabschwächung. Dieses wurde aber weder vom Kliniker erwähnt, noch sollte Clonazepam im Rahmen dieses TDM-Falls bestimmt werden.
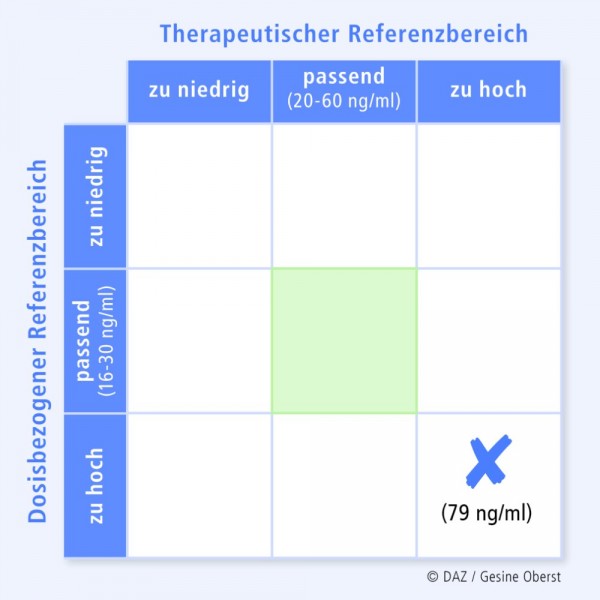
Fall 2
Eine 34-jährige, 108 kg schwere Patientin, Nichtraucherin, Kaffeetrinkerin, erhält 6 mg Risperidon täglich. Vereinzelt treten bei ihr Bewegungsstörungen auf. Im Blutplasma wurden eine Risperidonkonzentration von 61 ng/ml und eine Konzentration von 18 ng/ml für den aktiven Metaboliten 9-OH-Risperidon (syn. Paliperidon) gemessen. Die aktive Fraktion macht also 79 ng/ml aus, was oberhalb des therapeutischen Referenzbereiches liegt (Abb. 4). Die gemessene Plasmakonzentration liegt auch oberhalb des in Relation zur Dosis erwarteten Fensters (Dosis-bezogener Referenzbereich, Abb. 4).
Außer Risperidon erhält die Patientin noch zwei weitere Medikamente (Tab. 6). Risperidon hat mit Coffein, Erythromycin und Zopiclon gemeinsame Stoffwechselwege (CYP3A4/5); während Erythromycin die Metabolisierung über CYP3A4 hemmt, sind Coffein und Zopiclon in der Tabelle lediglich als Substrate dieser Enzyme verzeichnet.
|
Tab. 6: Aktuelle Medikation, Abbauwege und mögliche Interaktionen zu Fall 2 Modifiziert nach [4] sowie Fachinformationen | |||||
Wirkstoff |
CYP- |
||||
1A2 |
2C8/9 |
2C19 |
2D6 |
3A4/5 |
|
Risperidon |
X |
X |
|||
Erythromycin |
X H |
||||
Zopiclon |
X |
X |
|||
Coffein |
X H |
X |
X |
X |
|
X: Substrat/Eliminationsweg, H: Hemmung
Hauptabbauwege bei mehreren Eliminationswegen sind fett markiert. Dunkel hinterlegt ist die in diesem Fall als ursächlich erachtete Interaktion.Wie ist also diese erhöhte Plasmakonzentration zu erklären? Neben dem eher seltenen Fall einer Einnahme von mehr Risperidon als verordnet, scheint eine Enzymhemmung von CYP3A4 durch Erythromycin sehr wahrscheinlich.
Zu erwarten ist auch eine auf die Dosis bezogene erhöhte Plasmakonzentration von Zoplicon mit entsprechender Wirkverstärkung. Dieses wurde aber weder vom Kliniker erwähnt, noch sollte Zoplicon im Rahmen dieses TDM-Falls bestimmt werden.
Literatur
[1] Böhm R. Individualisierte Arzneimitteltherapie. In Herdegen (Hrsg). Kurzlehrbuch Pharmakologie und Toxikologie. Stuttgart 2010.
[2] Böhm R, et al. OpenVigil – free eyeballs on AERS pharmacovigilance data. Nat Biotechnol 2012; 30(2): 137 – 8.
[3] Evans SJ, et al. Use of proportional reporting ratios (PRRs) for signal generation from spontaneous adverse drug reaction reports. Pharmacoepidemiol Drug Saf 2001; 10(6): 483 – 6.
[4] Flockhart DA. Drug Interactions: Cytochrome P450 Drug Interaction Table. Indiana University School of Medicine, 2007; http://medicine.iupui.edu/clinpharm/ddis/table.aspx.
[5] Gonzalez FJ, et al. Evolution of the P450 gene superfamily: animal-plant ‚warfare‘, molecular drive and human genetic differences in drug oxidation. Trends Genet 1990; 6(6): 182 – 6.
[6] Greiner C. Interaktionslexikon Teil 6 – TDM – Die Kenngrößen. Neurotransmitter 2010; 5: 36 – 40.
[7] Haen E. Arzneimitteltherapiesicherheit/Pharmakovigilanz in der klinischen Psychopharmakotherapie. Psychopharmakotherapie 2011; 18: 238 – 43.
[8] Haen E. Der TDM-Befund. Psychopharmakotherapie 2012; 19: 129 – 34.
[9] Hiemke C, et al. AGNP consensus guidelines for therapeutic drug monitoring in psychiatry: update 2011. Pharmacopsychiatry 2011; 44(6): 195 – 235.
[10] Lin JH. CYP induction-mediated drug interactions: in vitro assessment and clinical implications. Pharm Res 2006;23(6): 1089 – 1116.
[11] Rendic S. Summary of information on human CYP enzymes: human P450 metabolism data. Drug Metab Rev 2002;34(1-2):83-448.
[12] Shimada T, et al. Interindividual variations in human liver cytochrome P-450 enzymes involved in the oxidation of drugs, carcinogens and toxic chemicals: studies with liver microsomes of 30 Japanese and 30 Caucasians. J Pharmacol Exp Ther 1994;270(1):414-23.
[13] Strandell J. Reporting patterns indicative of adverse drug interactions: a systematic evaluation in VigiBase. Drug Saf 2011;34(3):253-66.
Die Autoren danken Frau Dr. Petra Zagermann-Muncke (ABDATA) für Informationen über die ABDA-Interaktionsdatenbank.
Autoren
Dr. med. Ruwen Böhm, Dr. rer. nat. Kirstin Reinecke,
Prof. Dr. med. Dr. rer. nat. Ingolf Cascorbi,
Prof. Dr. med. Thomas Herdegen
Institut für Experimentelle und Klinische Pharmakologie
Arnold-Heller-Str. 3, Haus 30, 24105 Kiel
E-Mail: ruwen.boehm@pharmakologie.uni-kiel.de
Prof. Dr. med. Dr. rer. nat. Ekkehard Haen,
Abteilung Klinische Pharmakologie/Psychopharmakologie
Psychiatrische Universitätsklinik Regensburg Bezirksklinikum Regensburg,
Universitätsstraße 84, 93053 Regensburg
Der 1. Teil dieser Serie erschien in DAZ Nr. 36, Seite 64 – 74 unter dem Titel: "Arneimittelinteraktionen – verstehen, vermitteln und vermeiden".



0 Kommentare
Das Kommentieren ist aktuell nicht möglich.