















- DAZ.online
- DAZ / AZ
- DAZ 36/2012
- Arzneimittelinteraktionen
Klinische Pharmazie
Arzneimittelinteraktionen
Jede Pharmakotherapie ist mit dem Risiko unerwünschter Arzneimittelwirkungen (UAW) verbunden. Neben den prototypischen, durch den Wirkmechanismus verursachten UAW eines Medikamentes wie Müdigkeit bei Opioiden sind weitere wesentliche Ursachen von UAW:
- falsche Verordnung,
- falsche Applikation, Einnahmefehler, Übertragungsfehler,
- Arzneimittelinteraktionen sowie
- pharmakogenetische Besonderheiten.
Insbesondere ältere Patienten nehmen häufig mehrere – statistisch gesehen ca. fünf – Medikamente gleichzeitig ein. Die Anzahl der gleichzeitig verordneten Medikamente ist ein signifikanter Prädiktor für das Auftreten von UAW, verursacht durch Arzneimittelinteraktionen. Es ist daher notwendig, mögliche Arzneimittelinteraktionen zuverlässig und umfassend zu erkennen. Häufig werden UAW nicht als solche erkannt, sondern die Beschwerden werden als neues Symptom einer Erkrankung interpretiert.
Viele der Interaktionstypen, die in dieser Reihe geschildert werden, sind dank dem wachsenden Verständnis der molekularen Mechanismen und dem Vorliegen zahlreicher pharmakologischer Daten zu den zugelassenen Medikamenten vorhersagbar.
Mechanismen von Arzneimittelinteraktionen
Ein Arzneistoff wird bei oraler Gabe zunächst aus der Arzneiform freigesetzt, ein Vorgang dessen Geschwindigkeit von der Galenik des Arzneimittels geprägt ist. Der Arzneistoff wird dann resorbiert, im Körper verteilt und ggf. aktiviert (Pharmakokinetik, PK), bindet dann an seine Zielstruktur (Pharmakodynamik, PD), um anschließend über Niere oder Leber eliminiert zu werden (Pharmakokinetik) (Abb. 1).
Unter einer Arzneimittelwechselwirkung (syn. Arzneimittelinteraktion) wird eine Veränderung des Effektes durch die gleichzeitige Gabe eines anderen Arzneimittels verstanden. Derartige Arzneimittelwechselwirkungen sind in vielen Fällen erwünscht und erhöhen die Effektivität von Kombinationstherapien wie z. B. Kombination von Antihypertonika oder die Kombination von Analgetika und/oder Koanalgetika. Ebenso basiert die pharmakologische Behandlung einer Intoxikation auf dem Prinzip der Arzneimittelinteraktion bzw. Arzneimittel-Toxin-Interaktion wie z. B. Naloxon bei Opioidvergiftung oder Physostigmin bei Tollkirschenvergiftung.
Alle in Abbildung 1 gezeigten Prozesse können durch eine zweite Substanz, z. B. einen weiteren Arzneistoff, aber grundsätzlich auch durch andere Stoffe wie Hilfsstoffe in Arzneimitteln oder bestimmte Nahrungs- und Genussmittel, verändert werden. Wir unterscheiden vier Ebenen, auf denen Arzneimittelinteraktionen ablaufen können (Abb. 1, Beispiele in Tab. 1):
pharmakokinetische Interaktionen
– direkte, intermolekulare Interaktionen (physikochemische Inkompatibilitäten)
– indirekt, z. B. durch Veränderung der Aktivität von Transportern oder metabolisierenden Enzymen vermitteltpharmakodynamische Interaktionen
– direkt an derselben Zielstruktur
– indirekt über dasselbe Organ oder denselben Regelkreislauf
Tab. 1: Ausgewählte Interaktionen auf verschiedenen Ebenen | |||
Ebene |
Mechanismus (Auswahl) |
Interagierende Pharmaka (Auswahl) |
|
pharmako- kinetisch |
direkt, intermolekular (physikochemische Inkompatibilitäten) |
Chelatierung diverser Arzneistoffe durch polyvalente Kationen |
Calcium → Tetracyclin ↓1
Calcium → Bisphosphonate ↓
|
indirekt, z. B. über Enzyme |
Induktion oder Inhibition von arzneistoffmetabolisierenden Enzymen |
Hyperforin (Johanniskraut) (Induktor) → Nifedipin ↓
Clarithromycin (Inhibitor) → Nifedipin ↑
|
|
pharmako- dynamisch |
direkt (eine gemeinsame Zielstruktur) |
Kompetition an derselben Zielstruktur |
ASS + Ibuprofen
Salbutamol + Carvedilol
Morphin + Naloxon2
|
funktionell, indirekt |
Antagonismus oder Synergismus zweier Pharmaka im gleichen Regelkreislauf oder Erfolgsorgan |
Bisoprolol + Hydrochlorothiazid2
(Regelkreislauf: Blutdruck, erniedrigt) Dextromethorphan + Escitalopram
(Regelkreislauf: serotonerge Transmission, verstärkt) |
|
1 In dieser und folgenden Tabellen wird immer zuerst das Pharmakon aufgeführt, das die Wirkung des zweiten Pharmakons verändert. Hinter dem zweiten Pharmakon ist angegeben, in welche Richtung der Plasmaspiegel verändert wird (↑, ↓). Diese Unterscheidung gilt nur für pharmakokinetische Interaktionen.
2 Diese Wechselwirkungen sind i. d. R. erwünscht. Ein Antagonismus kann zu starke Wirkungen wie z. B. Atemdepression bei Opioidüberdosierung beenden (Antidot); ein Synergismus hilft dabei, die prototypischen UAW (z. B. Bradykardie durch hochdosierte Betablocker in der Monotherapie) zu begrenzen, während die erwünschte Wirkung wie z. B. Blutdrucksenkung verstärkt wird.Medikamente auf Interaktionen prüfen
Welche Möglichkeiten gibt es, schnell zwei oder mehr Medikamente auf Interaktionen zu überprüfen? Im täglichen Kundenkontakt ist der in den gängigen Apothekenkassenprogrammen integrierten Interaktions-Checker auf Grundlage der ABDA-Datenbank eine große Hilfe. Diese Datenbank kann auch über das Internet genutzt werden (Tab. 2).
Tab. 2: Interaktionsdatenbanken (Auswahl) | |
für Patienten | |
automatische, allgemeine Interaktions-Checker, zumeist ohne Angabe des Mechanismus, zumeist nur USAN, gute und einfache Bedienbarkeit |
|
automatischer, auf Anti-HIV-Medikamente spezialisierter Interaktions-Checker, ohne Angabe des Mechanismus und des klinischen Effekts, nur USAN |
|
für den wissenschaftlichen Gebrauch und – nach vorheriger individueller Prüfung – für den klinischen Gebrauch | |
Datenbank für CYP-basierte Interaktionen (mit Angabe der Primärliteratur) und Polymorphismen, auch Tabellen für Phase-2-Enzyme und Transporter, CYP-3D-Strukturen |
|
semi-manueller Interaktionscheck auf Basis der Daten der Drugbank, welche für viele Medikamente u. a. Zielstrukturen und metabolisierende Enzyme auflistet |
|
semiautomatischer Interaktions-Checker für CYP-Enzyme und den P-gp-Effluxtransporter |
|
http://www.agate-klinikverbund.de/downloads/ cyptabelle2301091.pdf |
Tabelle für Substrate, Induktoren und Inhibitoren von CYP-Enzymen, P-gp, UGT und renaler Transporter |
Tabelle für Substrate, Induktoren und Inhibitoren von CYP-Enzymen |
|
Tabellen für Substrate, Induktoren und Inhibitoren von SLC- und ABC-Transportern |
|
http://pharmacoclin.hug-ge.ch/activites_clinique/ pharma_outils.html |
diverse Anwendung und Übersichten für Interaktionen, nur französisch |
für den klinischen Gebrauch | |
http://medicine.iupui.edu/clinpharm/ddis/ ClinicalTable.aspx |
Tabelle für Substrate, Induktoren und Inhibitoren von CYP-Enzymen (s. o.) ; gekürzte Version nur für klinisch relevante Substanzen |
Interaktions-Check für Verordnungen mit Psychopharmaka, ausführliche Informationen zu Mechanismen, klinischer Relevanz , Handlungsempfehlungen und Links zu Literatur; kostenpflichtig
|
|
u. a. ABDA-Datenbank, kostenpflichtig
, zumeist ohne genaue Angabe des Mechanismus, u. a. mit weiteren Checks wie Dosisanpassung bei Niereninsuffizienz (DANI) |
|
Arzneimittelinformationsprogramm, kostenpflichtig
, neben weiteren Checks wie DANI auch ein Interaktionscheck |
|
CYP: Cytochrom-P450-Isoenzyme; ABC: ATP-binding cassette Effluxtransporter; DANI: Dosisanpassung bei Niereninsuffizienz; P-gp: P-Glykoprotein (ein ABC-Transporter); SLC: Solute carrier (Transporter); UGT: UDP-Glucuronosyl-Transferase; USAN: US adopted name
Die ABDA-Datenbank ist eine der umfangreichsten Datenbanken zu Interaktionen mit Schwerpunkt auf den in Deutschland aktuell und früher eingesetzten Medikamenten. Im März 2012 waren 427.941 Wechselwirkungen zwischen 1544 Stoffen und 31.207 Produkten (Fertigarzneimittel, Nahrungsergänzungsmittel, Diätetika, Medizinprodukte) hinterlegt. Die Datenbank wird 14-tägig aktualisiert. Die Warnungen enthalten immer Hinweise zum Schweregrad der Interaktion und häufig auch Empfehlungen zum weiteren Vorgehen wie z. B. Einnahmehinweise, Rücksprache mit dem Arzt, Verweigerung der Abgabe (Tab. 3). Teilweise wird der molekulare Mechanismus auch erklärt.
Tab. 3: Klassifikation der Schweregrade von Arzneimittelinteraktionen (AMI) und Umgangshinweise in der ABDA-Datenbank | |||
Kategorie |
Anteil der hinterlegten AMI |
Beispiel |
Hintergrund und Konsequenz |
schwerwiegende Folgen wahrscheinlich – kontraindiziert |
6% |
Simvastatin + Itraconazol |
in jedem Fall CYP3A4-vermittelt erhöhte Bioverfügbarkeit und damit Toxizität von Simvastatin
→ Rücksprache mit dem Arzt, Alternativen wie Pravastatin suchen
|
vorsichtshalber kontraindiziert |
11% |
Melatonin + Fluvoxamin |
laut Fachinformation kontraindiziert (theoretisch CYP1A2-vermittelt erhöhte Bioverfügbarkeit von Melatonin); keine In-vivo-Daten vorhanden;
aufgrund des ceiling-effect von Melatonin nicht klinisch relevant → Rücksprache mit Arzt, u. a. Info zur Arzthaftung beim Handeln gegen die Fachinformationen
|
Überwachung bzw. Anpassung notwendig |
46% |
Verapamil + Betablocker |
synergistische Wirkung mit Gefahr eines
AV-Blocks, insbesondere bei intravenöser Applikation des Betablockers → Applikationsweg und Dosis prüfen,
ggf. Info an Arzt |
in bestimmten Fällen Überwachung bzw. Anpassung notwendig |
4% |
ACE-Hemmer + NSAR |
in Abhängigkeit von der Einnahmedauer und der Dosis des NSAR Minderung der dauerhaften Blutdrucksenkung; Hyperkaliämie
→ Therapieregime erfragen und bei Vorliegen von Risikofaktoren Info an Arzt
|
vorsichtshalber überwachen |
31% |
Diazepam + Omeprazol |
laut Fachinformation Koadministration möglich, trotz verlangsamter Elimination von Diazepam wegen der Hemmung von CYP2C19; widersprüchliche In-vivo-Daten vorhanden
→ Info an Arzt
|
i. d. R. keine Maßnahme erforderlich |
2% |
Phenprocoumon + Hydrocortison |
in der Literatur wie z. B. Lehrbüchern wird die CYP-Induktion durch Glucocorticoide als möglich beschrieben, ist aber nicht klinisch relevant
(s. auch Dexamethason als Induktor von CYP3A4 in Tab. 5) → keine Maßnahme
|
Es gibt weitere Quellen, die auf einen oder einige wenige Interaktionsmechanismen spezialisiert und dadurch weniger umfangreich in Hinblick auf die Zahl der erfassten Medikamente und Interaktionen sind (Tab. 2). Dafür bieten sie im Regelfall aber sehr genaue Informationen mit Verweisen auf Literaturstellen.
Derzeit kann weder in deutschen Apotheken, noch in deutschen Krankenhäusern routinemäßig nach allen Interaktionen gesucht werden.
Die Apothekenkassenprogramme überblicken gegenwärtig nur den jeweils aktuellen Verkaufsvorgang, ggf. noch erweitert um Informationen zu früheren Einkäufen in der gleichen Apotheke, falls ein entsprechendes Einverständnis für die Aufnahme in die Kundendatei der jeweiligen Apotheke vorliegt. Pharmaka, die in unterschiedlichen Apotheken eingekauft worden sind, können ebenso wie freiverkäufliche Medikamente (z. B. aus Drogerien und aus Internetapotheken) für die Interaktionsprüfung nicht herangezogen werden. Besserung verspricht hier die elektronische Gesundheitskarte, die in Bochum-Wattenscheid im Rahmen des "Telematikinfrastruktur-unterstützte Erweiterung der Arzneimitteltherapiesicherheitsprüfungs-Datengrundlage als Mehrwertanwendung der elektronischen Gesundheitskarte (TEAM eGK)"-Projekts derzeit getestet wird. In anderen Ländern (z. B. Schweden) können alle Apotheker und Ärzte eine Liste aller rezeptierten (und herausgegebenen) Arzneimittel einsehen und so den Therapieverlauf und mögliche Interaktionen beurteilen.
In deutschen Krankenhäusern gibt es auch Pilotprojekte zur verbesserten Arzneimitteltherapiesicherheit: Am Universitätsklinikum in Kiel begleiten mittlerweile Stations- und Krankenhausapotheker die ärztlichen Visiten auf mehreren Stationen, optimieren z. B. die Arzneiform oder den Applikationszeitpunkt und warnen vor möglichen Interaktionen.
Praxis-TippEin an die ABDA-Datenbank angeschlossenes Apothekenkassenprogramm erkennt bei allen eingegebenen Medikamenten zuverlässig Arzneimittelinteraktionen und gibt Empfehlungen zum weiteren Vorgehen. Ggf. sollten freiverkäufliche Präparate und bestehende Medikation beim Verkauf mit eingegeben werden, um sie auch für die Prüfung zu berücksichtigen. Insbesondere bei Laufkunden muss neben der Dauermedikation standardisiert nach OTC-Präparaten wie NSAR, polyvalenten Kationen und Phytopharmaka gefragt werden. |
Erkennen von Arzneimittelinteraktionen
Arzneimittelinteraktionen wurden bereits seit den 1950er Jahren beschrieben: 1956 führte Axelrod die erste Studie zu Einflüssen auf den Morphinmetabolismus durch; aufbauend auf dem Konzept des Rezeptorantagonisten von Clark (1926), erstellte Dollery 1965 die erste ausführliche Betrachtung von Interaktionen mit Antihypertensiva; eine systematische Zusammenfassung mit Beispielen für verschiedene Interaktionstypen gab Morrelli 1968. Die Auftrittshäufigkeit von Interaktionen und damit ihre Bedeutung für den klinischen Alltag wurde aber erst später ersichtlich: Als Konsequenz aus der Thalidomid-Tragödie wurden 1969 in vielen Ländern Zentren zur Überwachung von Arzneimitteln auf der Basis einer spontanen Berichtserstattung, u. a. durch Ärzte und Apotheker, eingerichtet (sog. Pharmakovigilanz; Haen 2011; UAW-Meldung in Deutschland per www.abda.de/1087.html). Schnell wurden in diesen Pharmakovigilanzdaten auch Arzneimittelinteraktionen beobachtet (s. Infobox OpenVigil), deren molekulare Mechanismen jedoch erst Jahre bis Jahrzehnte später aufgeklärt werden konnten oder sogar immer noch unbekannt sind (Abb. 2).
OpenVigilEin Instrument aus der Pharmakologie in Kiel zur Analyse von Interaktionen aus PharmakovigilanzdatenDie meisten Pharmakovigilanzzentren veröffentlichen ihre Daten nicht, sondern führen Analysen, die z. B. zur Entdeckung einer Arzneimittelinteraktion führen können, selbst durch. Gründe dafür sind u. a. die besonderen Limitationen dieser Daten (www.uni-kiel.de/pharmacology/pvt/caveat.html). Die Pharmakovigilanzdaten der FDA sind der Öffentlichkeit zugänglich und können z. B. durch das von uns entwickelte OpenVigil-Analyseprogramm eingesehen werden (www.uni-kiel.de/pharmacology/pvt; Nat Biotechnol 2012 Feb 8;30(2):137-8). Die FDA codiert unerwünschte Ereignisse (UE) nach dem MedDRA-Wörterbuch. Für Interaktionen sind z. B. folgende unerwünschte Ereignisse mit jeweiligen Häufigkeiten hinterlegt (Pharmakovigilanzdaten sind vom 1. Quartal 2004 bis zum 3. Quartal 2011, ab 400 Fällen):
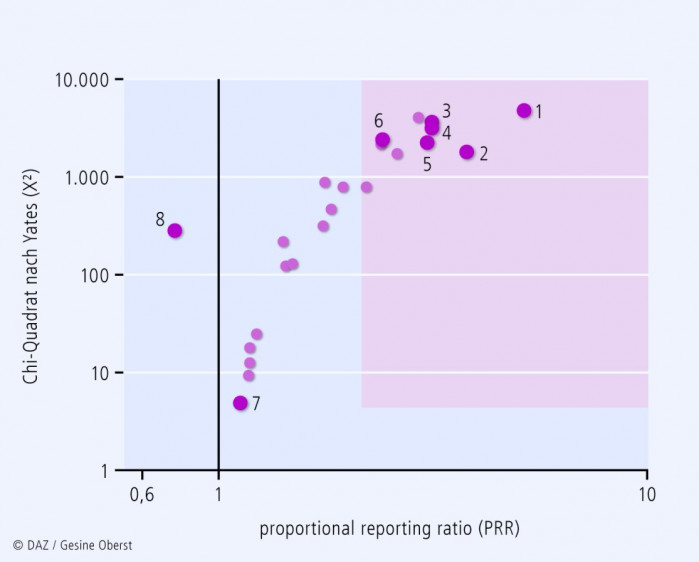
Sucht man für "Drug interaction" nach den beteiligten Medikamenten, ergibt sich folgende Liste (nur Medikamente, benannt nach verschiedenen Nomenklaturen, mit mehr als 1250 gemeldeten Interaktionen; cave die Verzerrung durch alleinige Betrachtung bekannter oder vermuteter Interaktionen!): Aspirin 3450 Simvastatin 2559 Warfarin Sodium 2443 Omeprazole 2122 Furosemid 2018 Seroquel 1603 Lyrica 1495 Levothyroxine sodium 1367 Atenolol 1356 Ramipril 1336 Lipitor 1326 Zocor 1304 Chantix 1301 Das die Liste anführende Medikament "Aspirin" (INN: Acetylsalicylsäure) führt übrigens nur numerisch aufgrund des weitverbreiteten Einsatzes; tatsächlich ergibt sich nach genauerer statistischer Auswertung (Berechnung der Proportional Reporting Ratio (PRR), Abb. 3) keine signifikante Wahrscheinlichkeit, dass ASS überhäufig an Interaktionen beteiligt ist. Für diese Analyse wurde jedoch nicht nach unterschiedlichen Dosierungen stratifiziert. Auf Basis einer für INN, USAN, Markennamen und Salze bereinigten Liste wurde das Diagramm in Abb. 3. erstellt, in dem die am häufigsten interagierenden Medikamente aufgeführt sind. |
Nr. |
Medikament |
Typ der Interaktion |
häufige Interaktions-bedingte UAW/Bemerkung |
1 |
Clarithromycin |
PK |
CYP3A4-Inhibition verstärkte Wirkung zahlreicher Arzneimittel |
2 |
Escitalopram |
PD |
Tremor, Blutungen |
3 |
Atenolol |
PD |
Bradykardie |
4 |
Simvastatin |
PK |
CYP-Substrat Rhabdomyolyse durch erhöhte Plasmaspiegel |
5 |
Ramipril |
PD |
kardiale und renale Komplikationen; Kausalität jedoch fraglich und Schweregrad eher gering |
6 |
Warfarin |
PK |
CYP2C9-Substrat Blutungen durch erhöhte Plasmakonzentrationen |
7 |
ASS |
ASS und Paracetamol stehen aufgrund der zahlreichen Verordnungen oben in der Liste häufig interagierender Pharmaka. Tatsächlich sind sie jedoch nicht häufiger problematisch als alle anderen Medikamente |
|
8 |
Paracetamol |
Die US-amerikanische Zulassungsbehörde FDA veröffentlichte 1997 ihre erste Empfehlung, wie vor der Zulassung eines neuen Arzneistoffes auf die wichtigsten Interaktionsmechanismen geprüft werden kann. So wird heute bereits in der präklinischen und klinischen Entwicklung das Interaktionspotenzial eines Arzneistoffs aufgrund seines Verhaltens an zahlreichen metabolisierenden Enzymen und Aufnahme- und Efflux-Transportern charakterisiert. Verschiedene Modelle kommen hier zum Einsatz und ergänzen sich komplementär. Die beste Aussagekraft haben dabei humane In-vivo-Studien, da ansonsten z. B. posttranskriptionale Effekte oder indirekte pharmakodynamische Interaktionen nicht erfasst werden können.
Auch heute noch werden nach der Zulassung bis dato nicht erfasste Arzneimittelinteraktionen bei der Analyse von Pharmakovigilanzdaten beobachtet (s. Infobox OpenVigil). Mit steigenden und immer komplexeren Anforderungen für die Zulassung neuer Arzneimittel, liegen Hinweise auf viele potenzielle Interaktionen bereits vor der Marktzulassung vor. So war z. B. auch die in einigen Fällen tödlich verlaufende Interaktion von Cerivastatin (Lipobay®) mit Gemfibrozil bei der Zulassung bekannt. Leider wurde sie nicht von allen verschreibenden Ärzten und abgebenden Apothekern in den USA beachtet.
Häufige Arzneimittelinteraktionen
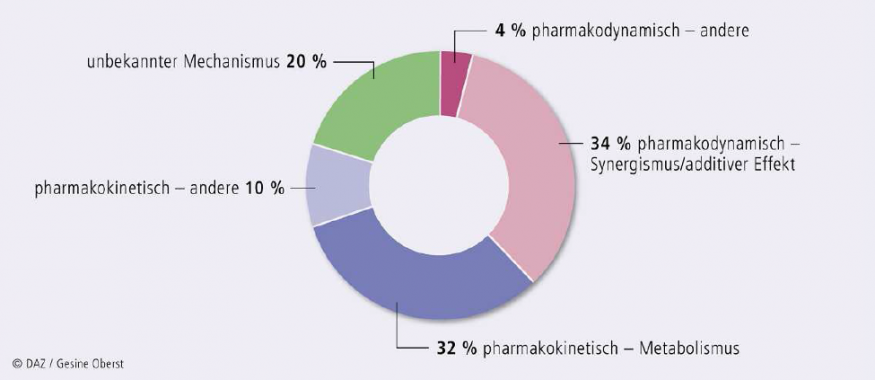
Die oben beschriebenen Auswertungen von Pharmakovigilanzdatenbanken erkennen Arzneimittelinteraktionen und an Interaktionen beteiligte Medikamente. Die Ergebnisse einer von uns auf der Basis der US-amerikanischen FDA-Pharmakovigilanzdaten durchgeführten Risikoanalyse sind in Abbildung 3 dargestellt: Strandell vom WHO Pharmakovigilanzzentrum publizierte 2011 eine Übersicht über die häufigsten Interaktionstypen. Auffällig ist, dass das Gros der Meldungen (86%) sich auf durch die Interaktion verstärkte Toxizität von Arzneistoffen bezieht. Wirkungsverluste, bedingt durch Interaktionen, wurden wesentlich seltener gemeldet. Tatsächlich fallen toxische Effekte klinisch stärker auf. Wirkungsverluste hingegen sind nur bei einigen besonderen Medikamentengruppen problematisch (z. B. Transplantatabstoßung bei Immunsuppressiva oder Resistenzbildung bei unzureichenden Plasmakonzentrationen und damit unzureichender antimikrobieller Wirkung von Antibiotika). Weiterhin zeigen Abb. 2 und 3, dass am häufigsten die indirekten, funktionellen pharmakodynamischen Interaktionen auftreten, dicht gefolgt von indirekten pharmakokinetischen Interaktionen, v. a. bedingt durch Veränderung von Enzymaktivitäten. In ca. 20% der beobachteten Interaktionen ist der genaue Mechanismus immer noch unbekannt.
Umgang mit Arzneimittelinteraktionen
Entscheidend für eine Bewertung von Arzneimittelinteraktionen sind Dauer und Schwere der Interaktion, die Darreichungsform sowie die therapeutische Breite der betroffenen Medikamente.
Interaktionen durch ein neues Medikament
Wenn ein neues Medikament zu einem bestehenden Therapieregime bzw. zu einer Ernährung mit interagierenden Xenobiotika (Grapefruit) oder Elektrolyten (Natrium) hinzugegeben wird, kann es innerhalb von Minuten (z. B. bei i.v.-Injektion) bis Tagen/Wochen (s. Infobox Der Zeitfaktor) zu potenziellen Wechselwirkungen kommen. Bei schwerwiegenden Interaktionen (z. B. extreme Blutdrucksenkung durch Nifedipin, Atemstillstand durch Fentanyl) kann im Krankenhaus der Arzt sofort entsprechend korrigierend eingreifen. Ambulante Patienten ohne sofortige ärztliche Hilfe sind einem größeren Risiko ausgesetzt. An dieser Stelle sind die Apotheken gefragt. Hier laufen häufig die Informationen zusammen und mittels gezielter Beratung können zahlreiche Interaktionen vermieden werden.
Der ZeitfaktorMechanismus und HWZ entscheiden über Beginn und DauerFür die Bewertung, wie schnell eine mögliche Arzneimittelinteraktion auftritt und anhält, sind die Halbwertzeiten der interagierenden Medikamente sowie der Interaktionsmechanismus entscheidend:
|
Wie kann nun die praktische Umsetzung in der Apotheke aussehen? Wann muss gehandelt werden?
Bei allen (laut ABDA-Datenbank, s. Tab. 3) als schwerwiegend eingestuften Interaktionen muss eingegriffen werden. Hier ist bei rezeptierten Arzneimitteln ein Anruf beim Arzt unabdingbar. Bei mittelschweren Interaktionen wird abgewogen. Die Kombination führt zu therapeutischen Schwierigkeiten, doch bei sorgfältiger Überwachung des Patienten (z. B. INR-Werte während einer Behandlung mit Phenprocoumon, klinische Symptome bei einer Behandlung mit Neuro- und Psychopharmaka) kann die Kombination verabreicht werden. Dabei ist zu berücksichtigen, welches Arzneimittel zu welchem bestehenden Behandlungsregime hinzukommt, ob es sich um eine kurzfristige Einnahme handelt wie z. B. den kurzzeitigen Einsatz von NSAR bei bestehender Antikoagulation mit ASS oder den kurzzeitigen Einsatz von NSAR bei antihypertensiver Therapie. Auch die gewählte Darreichungsform bestimmt den Schweregrad der Interaktion, denn bei oraler Applikation fallen pharmakokinetische Interaktionen aufgrund des First-pass-Metabolismus stärker ins Gewicht. Hier kann schon eine zeitlich versetzte Einnahme Abhilfe schaffen wie bei z. B. Calcium und Bisphosphonaten oder ASS und Ibuprofen. Bei Selbstmedikation kann auch ein Alternativpräparat empfohlen werden wie z. B. Pantoprazol statt Omeprazol. Aber Vorsicht bei irreversibler Enzymhemmung wie z. B. durch Grapefruitsaft, Enzyminduktion oder interagierende Medikamente mit langer HWZ (s. Infobox Der Zeitfaktor)! Denn hier stellt eine zeitlich versetzte Einnahme keine Alternative dar. Bei als geringfügig eingestuften Wechselwirkungen können etwas verstärkte bzw. verminderte Wirkungen auftreten, oder aber sie betreffen nur einen bestimmten Personenkreis mit Risikofaktoren. Hier muss dann das weitere Vorgehen individuell entschieden werden.
Eine generelle Beratung, wie Zeichen einer UAW erkannt werden können, darf nicht fehlen. Ein gut informierter Patient weiß, wie man auf UAW (z. B. Blutungen unter Phenprocoumon, Hypoglykämie unter Antidiabetika und Betablockern, Zwischenblutungen bei hormoneller Kontrazeption) reagieren muss und kann so das Ausmaß dieser UAW minimieren.
Praxis-Tipp Häufige InteraktionenDer häufigste Interaktionstyp ist die durch Abbauhemmung oder Synergismus hervorgerufene Toxizität eines Arzneistoffs. Insbesondere bei Medikamenten mit geringer therapeutischer Breite und bei Medikamenten mit bekanntem Interaktionsrisiko (s. Abb. 3 und Tab. 3) sollte der Kunde geschult werden, typische Überdosierungseffekte zu erkennen (z. B. Hämatome durch Gerinnungshemmer, Muskelschmerzen durch Statine). Der weniger häufige Fall eines Wirkungsverlustes ist insbesondere bei Antibiotika, Immunsuppressiva und Gerinnungshemmern kritisch. |
Arzneimittelinteraktionen durch Absetzen
Ist eine Arzneimittelinteraktion durch entsprechende Dosisanpassungen austariert, besteht die Gefahr, dass nach Absetzen eines Medikamentes erneut Probleme auftreten. Beim Absetzen von Inhibitoren oder Induktoren von Arzneimittel-metabolisierenden Enzymen sind gegenläufige Effekte auf die Plasmakonzentration denkbar (sog. Deinhibition bzw. Deinduktion). Neben Medikamenten können auch Xenobiotika aus der Nahrung und Elektrolyte bei Veränderung der Konsumgewohnheiten Probleme verursachen (z. B. Clozapintoxizität durch plötzliche Nicotinabstinenz, Lithiumtoxizität durch plötzliche salzarme Ernährung).
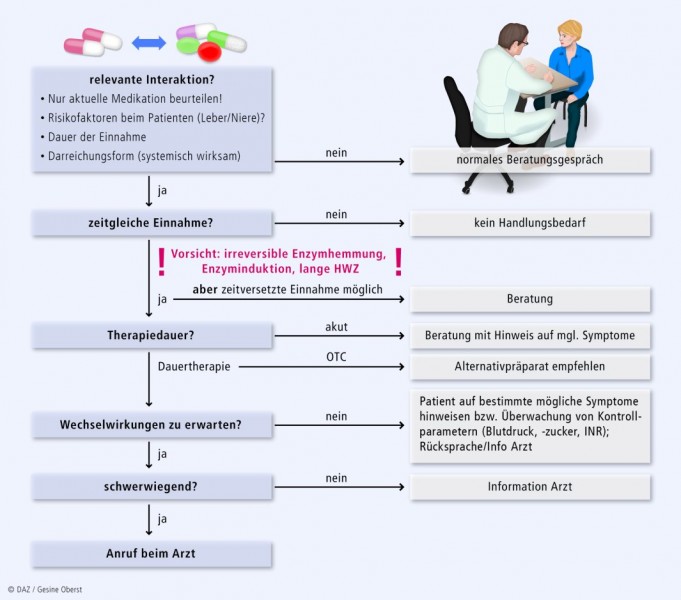
Ein Algorithmus, wie man schnell die o. g. Verfahrensweisen umsetzen kann, ist in Abbildung 4 dargestellt.
Anzuraten wäre auch das Etablieren eines apothekeneigenen Interaktionsmanagements anhand der Abverkaufszahlen von Schnelldrehern mit erhöhtem Interaktionspotenzial.
Pharmakokinetische Interaktionen
Im Bereich der Pharmakokinetik sind Arzneimittelinteraktionen auf folgenden mechanistischen Ebenen denkbar (Tab. 4):
- Absorption,
- Distribution,
- Metabolismus (= Giftung und Entgiftung) und
- Exkretion.
Tab. 4: Ausgewählte Interaktionen auf pharmakokinetischer Ebene | ||
Ebene |
Mechanismus |
interagierende Pharmaka (Auswahl) |
Absorption |
Veränderung der GIT-Passagezeit |
Laxanzien (Lactulose), Opioide (Codein), Anticholinergika (Butylscopolamin) → veränderte AUC, Cmax
, tmax
anderer Medikamente |
pH-Veränderung |
Antacida → Pseudoephedrin ↑
Antacida oder Calciumcarbonat → Schilddrüsenhormone ↓
|
|
Emulgierung |
fettreiche Nahrung → Saquinavir ↑
Colestyramin → Östrogene ↓
|
|
Induktion von intestinalen Effluxtransportern |
Rifampicin → Digoxin ↓ |
|
Distribution |
Hemmung von Aufnahmetransportern |
Gemfibrozil → Cerivastatin1
(Lipobay©) Toxizität ↑, Wirkung ↓
Penicillin → Amanitin (Knollenblätterpilz) Toxizitität ↓
|
Metabolismus |
Induktion oder Inhibition arzneistoffmetabolisierender Enzyme, z. B. CYP |
Hyperforin aus Johanniskraut (Induktor) → Ciclosporin ↓
Verapamil (Inhibitor) → Ciclosporin ↑
|
Exkretion |
(Kompetitive) Hemmung von Aufnahme- und Effluxtransportern |
ASS → Methotrexat ↑ |
Einleitend werden zunächst alle denkbaren Ebenen von pharmakokinetischen Arzneimittelinteraktionen zusammengefasst und die Familie der Cytochrom-P450-Enzyme (CYP) vorgestellt. Dann werden pharmakokinetische Interaktionen am Beispiel des Arzneistoff-metabolisierenden Isoenzyms CYP3A4 vorgestellt.
Ziel des Arzneistoffmetabolismus, dem ca. 65% aller gängigen Medikamente unterliegen, ist die Verbesserung der Löslichkeit eines Xenobiotikums (= körperfremde Substanzen), so dass es leichter renal eliminiert werden kann. Beim Metabolismus von Arzneistoffen unterscheidet man Phase I und II-Reaktionen. Durch Phase-I-Enzyme der Cytochrom-P450-Familie wird durch Redox-Reaktionen wie Hydrolyse, Desalkylierung, Desaminierung oder Oxidation eine funktionelle Gruppe in das Xenobiotikum eingebracht oder abgespalten. Phase-II-Enzyme transferieren ein weiteres Molekül wie Methylgruppen, Acetylgruppen, Sulfat, Glycin oder Glutathion auf das Xenobiotikum. Trotz dieser Nomenklatur und Einteilung in Phase I und Phase II müssen diese Reaktionen nicht zusammen, sondern können auch unabhängig voneinander oder in umgekehrter Reihenfolge erfolgen.
Metabolismus führt nicht in allen Fällen zur Entgiftung eines Xenobiotikums. Es kann auch eine Aktivierung (syn. Giftung) eines sog. Prodrugs erfolgen (z. B. das wenig wirksame Codein zum sehr wirksamen Morphin), oder es können ähnlich der Muttersubstanz aktive Metaboliten entstehen (z. B. Amitriptylin zu Nortriptylin).
Praxis-TippInteraktionen vorbeugenDurch Arzneimittelinteraktionen verursachte UAW treten nach Veränderungen der Medikation oder nach Veränderung von Konsumgewohnheiten auf. In den ersten Stunden bis Tagen nach An- oder Absetzen eines Medikamentes sollte daher verschärft auf UAW oder Wirkungsverlust geachtet und der Patient entsprechend sensibilisiert werden. Patienten müssen darauf hingewiesen werden, dass auch die Veränderung von bestimmten Lebensgewohnheiten wie Rauchen, Trinken von Grapefruitsaft oder salzreiche Ernährung bei ihnen trotz gut eingestellter Medikation neue ernsthafte Probleme verursachen kann. Ist eine Interaktion bekannt, gibt die ABDA-Datenbank Empfehlungen zum weiteren Vorgehen (Tab. 3, Abb. 4). |
Cytochrom-P450-Enzyme
Cytochrom-P450-Isoenzyme sind – neben Transportern – das wichtigste Verteidigungssystem unseres Körpers gegen körperfremde Stoffe (Xenobiotika) wie die potenziell giftigen sekundären Pflanzenstoffe oder Arzneimittel. Sie werden mit den Buchstaben "CYP", der Nummer ihrer Familie (> 40% Homologie), dem Buchstaben der Subfamilie (> 55% Homologie) und einer Nummer abgekürzt (z. B. CYP3A4). Insbesondere Isoenzyme der (Sub‑)Familien CYP1A, CYP2 und CYP3A sind für die Aktivierung und Entgiftung der heute eingesetzten Arzneistoffe und anderer Xenobiotika essenziell.
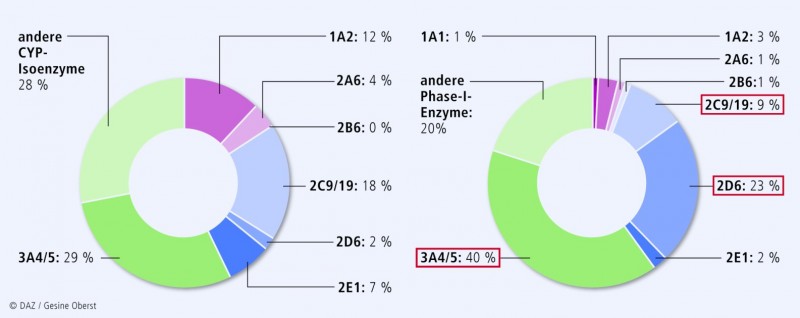
Cytochrom-P450-Enzyme werden besonders stark in der Leber und in den Enterozyten des Intestinums exprimiert (Expressionsmuster der Isoenzyme s. Abb. 5). Arzneimittelinteraktionen auf CYP-Ebene sind im Allgemeinen ausgeprägter, je höher der First-pass-Effekt ist. Aber auch nach parenteraler Verabreichung von Pharmaka kann dieser Interaktionstyp klinisch relevant sein. Als Phase-I-Enzyme metabolisieren CYP über die Hälfte der auf dem Markt befindlichen Medikamente. Die meisten klinisch relevanten Substrate hat dabei CYP3A4 (Abb. 5).
Manchmal wird ein Substrat durch mehre CYP verstoffwechselt, was das Interaktionspotenzial minimieren kann. Ein Beispiel ist das Opioid Oxycodon, welches zwar über CYP2D6 und CYP3A4 und vermutlich noch weitere CYP verstoffwechselt wird, jedoch in seiner Pharmakokinetik bei isolierter Hemmung oder Induktion eines CYP nicht klinisch relevant beeinträchtigt wird (CYP-neutral).
Die Beteiligung verschiedener CYP an der Verstoffwechslung eines Pharmakons kann sich konzentrationsabhängig verändern: So wird Mirtazapin bei niedrigen Plasmakonzentrationen primär von CYP2D6 hydroxyliert. Bei höheren Konzentrationen überwiegt die CYP3A4-vermittelte N-Demethylierung.
CYP3A4 wird hepatisch stark exprimiert und besitzt das breiteste Substratspektrum. Es stellt somit das – insbesondere bei hohen Konzentrationen toxischer Xenobiotika – wichtigste Abwehrsystem unseres Körpers gegen Xenobiotika dar. (Rohdaten aus Shimada, 1994, und Rendic, 2002)
CYP-Aktivität und Polymorphismen
Cytochrom-P450-Enzyme können polymorph exprimiert sein: Es gibt Unterschiede in der Metabolisierungsgeschwindigkeit. Bezüglich des Metabolisiererstatus unterscheidet man "normale" Metabolisierer (extensive metabolizer, EM), also den häufigsten Metabolisiererstatus, an den auch die empfohlene Dosierung von Arzneimitteln angepasst wird, sowie besonders langsame (poor metabolizer, PM) und bei einigen Enzymen auch besonders schnelle Metabolisierer (ultrarapid metabolizer, UM). In anderen Populationen und/oder bei einigen Enzymen können auch PM oder UM der häufige Normalfall sein.
Das Verhältnis von Ausgangssubstanz zu Metabolit, die sog. metabolic ratio, kann dabei helfen, die Metabolisierungsgeschwindigkeit abzuschätzen (Hiemke 2011, Haen 2012). Die metabolic ratio erlaubt allerdings keine Unterscheidung, ob ein Polymorphismus, eine Arzneimittelinteraktion oder Non-Compliance ursächlich für den abweichenden Wert ist.
Eine Veränderung der CYP-Metabolisierungsgeschwindigkeit – unabhängig davon, ob angeboren oder durch Komedikation verursacht – kann insbesondere bei Medikamenten mit geringer therapeutischer Breite (z. B. Schilddrüsenhormone, Herzglykoside, Sulfonylharnstoffe, Gerinnungshemmer, Calciumkanalblocker, trizyklische Antidepressiva) ausgeprägte klinische Konsequenzen haben (Abb. 6).
Praxis-TippVon pharmakokinetischen Arzneimittelwechselwirkungen sind insbesondere lipophile Pharmaka betroffen, die häufig von CYP für die Ausscheidung vorbereitet werden müssen. Zu diesen lipophilen Pharmaka, die auch teilweise als Inhibitor wirken, zählen v. a. solche, welche in tiefe Kompartimente eindringen, wie
|
CYP-Aktivität – Inhibitoren und Induktoren
Durch Komedikation können CYP-Enzyme induziert (durch nukleäre Rezeptoren verstärkte Expression, Stabilisierung von mRNA oder Protein, kooperative Effekte) oder inhibiert (kompetitive oder nicht-kompetitive, irreversible Hemmung) werden (Tab. 5). Von diesen genannten Mechanismen sind vor allem die gesteigerte Genexpression und die Enzymhemmung klinisch relevant.
Tab. 5: Mechanismen, die die Aktivität von Cytochrom-P450-Enzymen verändern | |||
Mechanismus |
Zielstruktur |
Beispiel(e) |
klinischer Effekt |
Induktion = verstärkte CYP-Aktivität | |||
gesteigerte Gentranskription durch Stimulation nukleärer Rezeptoren |
GR (darüber vermehrte PXR-Expression) |
Dexamethason – CYP3A |
erhöhte Lebertoxizität durch toxische Metaboliten des CYP3A4-Substrates Lapatinib in vitro (in vivo noch nicht ausreichend untersucht) |
AhR |
Polycyclische aromatische Kohlenwasserstoffe (z. B. Benzo[a]pyren im Rauch) – CYP1A |
klinisch bedeutsam verstärkte Entgiftung von CYP1A2-Substraten wie Clozapin bei Rauchern |
|
CAR |
Hyperforin - CYP3A4 |
klinisch bedeutsam verstärkte Entgiftung von CYP3A4-Substraten wie Ciclosporin A |
|
PXR | |||
Hemmung des Molekülabbaus |
mRNA |
Nahrungskarenz, Diabetes mellitus – CYP2E1-mRNA |
keiner bekannt |
Protein |
Alkohole und Ketone wie Ethanol, Isoniazid – CYP2E1 |
erhöhte Lebertoxizität von Paracetamol; klinisch bedeutsam beschleunigte Entgiftung von CYP2E1-Substraten wie Isofluran |
|
kooperative Effekte |
Enzym/ Transporter |
Diclofenac oder Warfarin und Chinidin – CYP3A4; beschrieben in vitro und in vivo (Affe) |
keiner bekannt |
Inhibition = abgeschwächte CYP-Aktivität | |||
kompetitive Hemmung |
Protein |
Chinidin – CYP2D6 |
klinisch bedeutsam verlangsamte Entgiftung von CYP2D6-Substraten wie Metoprolol |
nicht-kompetitive, meist irreversible Hemmung |
Paroxetin – CYP2D6 |
||
Medikamentengruppen, bei denen mit CYP-Interaktionen gerechnet werden muss, sind vor allem lipophile Pharmaka. Sie dringen gut in Gewebe (z. B. Antiinfektiva) oder ZNS (z. B. Neuropharmaka) ein. Sie müssen daher durch CYP hydrophiler gemacht und zur renalen Ausscheidung vorbereitet werden. Viele Medikamente sind dabei nicht nur Substrate der CYP, sondern wirken gleichzeitig auch als Induktor oder Inhibitor.
Autoren
Dr. med. Ruwen Böhm, Dr. rer. nat. Kirstin Reinecke, Prof. Dr. med. Dr. rer. nat. Ingolf Cascorbi, Prof. Dr. med. Thomas Herdegen, Institut für Experimentelle und Klinische Pharmakologie, Arnold-Heller-Str. 3, Haus 30, 24105 Kiel
Prof. Dr. med. Dr. rer. nat. Ekkehard Haen, Abteilung Klinische Pharmakologie/Psychopharmakologie Psychiatrische Universitätsklinik Regensburg Bezirksklinikum Regensburg, Universitätsstraße 84, 93053 Regensburg



0 Kommentare
Das Kommentieren ist aktuell nicht möglich.