


















- DAZ.online
- News
- Pharmazie
- Neue EU-Medizinprodukte-...
MDR
Neue EU-Medizinprodukte-Verordnung: Was ändert sich für Apotheken?
Berlin - 24.05.2019, 16:15 Uhr

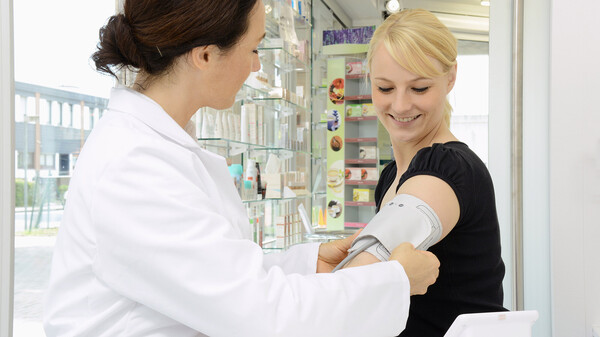
Ist das Medizinprodukt korrekt gekennzeichnet? Mit der neuen Medizinprodukte-Verordnung kommen auf Apotheken demnächst neue Prüf- und Dokumentationspflichten zu. (s / Foto: imago images / blickwinkel)
Vorsicht beim Umpacken
Weitere Neuerungen betreffen Händler „mit besonderen Tätigkeiten“ also Händler, die Änderungen an Medizinprodukten oder deren Verpackung vornehmen. Hier erhöht sich der Aufwand für Apotheken allerdings erheblich. Denn Apotheken müssen den Hersteller sowie die zuständige Behörde 28 Tage vor der geplanten Abgabe des veränderten Medizinprodukts informieren. Außerdem müssen Produkte, deren Verpackung geändert wurde, mit Namen und Adresse der Apotheke gekennzeichnet und der Vorgang im QMS-Handbuch dokumentiert werden.
Kombiniert die Apotheke unterschiedliche Medizinprodukte in einer neuen Verpackung – ein Beispiel ist das Zusammenstellen von OP-Sets für Praxen, die ambulante Eingriffe durchführen –, bringt die MDR zusätzliche Anforderungen mit sich: Und zwar muss die Apotheke nachweisen, dass die Einzelkomponenten miteinander physikalisch kompatibel sind, auch wenn sie gemeinsam sterilisiert werden. Diese „Beweisführung“ dürfte die Kapazitäten der meisten Apotheken allerdings überschreiten.
Leichter wird es dagegen bei Kompressionsstrümpfen nach Maß. Diese fallen gemäß MDR nicht mehr unter die Kategorie der Sonderanfertigungen, mit denen verstärkte Dokumentationspflichten verbunden sind.
„Marktbereinigung möglich“
Für Inverkehrbringer von Medizinprodukten ist der Mehraufwand, der durch die MDR entsteht, noch größer als für Apotheken. So gibt es Änderungen bei den Klassifizierungsregeln, die überwiegend mit einer Höherklassifizierung verbunden sind. „Dies könnte zu einer Marktbereinigung führen“, prognostiziert Ertner.
Aus Sicht des
Medizinprodukte-Experten tragen die neuen Regelungen zur Patientensicherheit
bei. „Insbesondere die verstärkten Meldepflichten aller Wirtschaftsakteure und
die Verpflichtung zur Nachbeobachtung von Medizinprodukten über den gesamten
Produktlebenszyklus durch die Hersteller bringen künftige Verbesserungen bei
der Sicherheit von Medizinprodukten mit sich“, findet Ertner. Allerdings hat er
Bedenken, ob das Timing für die nationale Umsetzung eingehalten werden kann: „Nach
Informationen des Bundesverbandes Medizintechnik BVMed hat derzeit nur eine
einzige der 57 Benannten Stellen in der EU die Neubenennung für die MDR
abgeschlossen, weniger als die Hälfte konnte bisher auditiert werden. Hinzu kommt,
dass ein Teil der Benannten Stellen ihren Sitz in Großbritannien hat. Für die
zentrale europäische Datenbank Eudamed gibt es noch viele offene Punkte zu
klären, bis diese in der Lage ist, die Aufgaben der MDR zu erfüllen. Der
Umsetzungstermin Ende Mai 2020 wird denkbar knapp werden.“



10 Kommentare
Medizinprodukte-Verordnung
von Uto Ziehn am 01.12.2019 um 14:33 Uhr
» Auf diesen Kommentar antworten | 0 Antworten
Frage von Herrn Thomas Witschel am 04.08.2019
von Dr. Gert Schorn am 06.08.2019 um 16:42 Uhr
» Auf diesen Kommentar antworten | 0 Antworten
Auseinzeln
von Christoph am 26.05.2019 um 12:55 Uhr
» Auf diesen Kommentar antworten | 2 Antworten
AW: Auseinzeln
von Dr. Gert Schorn am 27.05.2019 um 12:11 Uhr
AW: Auseinzeln
von Dr. Gert Schorn am 27.05.2019 um 12:16 Uhr
Kommentar von Heilo Barz
von Dr. Gert Schorn am 25.05.2019 um 17:33 Uhr
» Auf diesen Kommentar antworten | 1 Antwort
AW: Kommentar von Heilo Barz
von Thomas Witschel am 04.08.2019 um 8:43 Uhr
Diktatur der EURO-Richtlinien und Gesetze!
von Heiko Barz am 25.05.2019 um 11:33 Uhr
» Auf diesen Kommentar antworten | 0 Antworten
Neues EU-Recht zu Medizinprodukten und In-vitro-Diagnostica
von Dr. Gert Schorn am 24.05.2019 um 22:43 Uhr
» Auf diesen Kommentar antworten | 1 Antwort
AW: Neues EU-Recht zu Medizinprodukten und
von Bettina Jung am 27.05.2019 um 9:48 Uhr
Das Kommentieren ist aktuell nicht möglich.