

















- DAZ.online
- News
- Pharmazie
- Hamburger ...
Umstrittener Valsartan-Hersteller
Hamburger Arzneimittelaufsicht inspizierte Zhejiang Huahai regelmäßig
Stuttgart - 24.09.2018, 14:45 Uhr

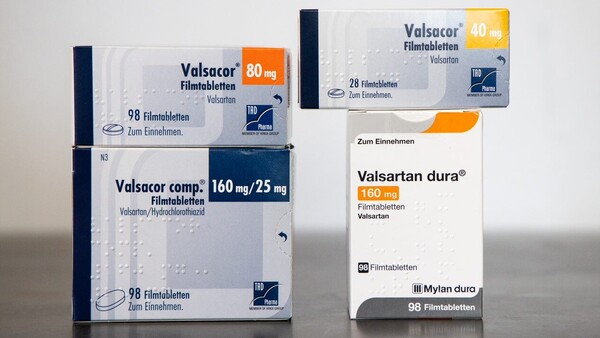
Der chinesische Hersteller Zhejiang Huahai, der das verunreinigte Valsartan produzierte, wurde in den vergangenen Jahren auch mehrfach von einer deutschen Behörde inspiziert. (s / Symbolbild, Foto: picture alliance)
Dass die US-amerikanische Arzneimittelbehörde FDA schon 2017 auf eine unbekannte Verunreinigung beim chinesischen Valsartan-Hersteller Zhejiang Huahai aufmerksam wurde, ist bekannt. Um welchen Wirkstoff es dabei ging, weiß man nicht. In den vergangenen Jahren war aber auch eine deutsche Behörde regelmäßig bei Zhejiang Huahai und führte Inspektionen durch. DAZ.online hat bei der Behörde für Gesundheit und Verbraucherschutz aus Hamburg nachgefragt, was es mit ihren Inspektionen auf sich hatte.
Der blutdrucksenkende Wirkstoff Valsartan hat im Sommer 2018 weltweit traurige Berühmtheit erlangt. Berühmt gemacht haben ihn zwei Verunreinigungen, die durch eine Umstellung der Synthese im Jahr 2012 in den Wirkstoff gelangt sein könnten – die beiden Nitrosamine NDMA und NDEA, die wahrscheinlich krebserregend sind. Im Mittelpunkt des Skandals steht der chinesische Wirkstoffhersteller Zhejiang Huahai, aber auch ein Wirkstoffhersteller aus Indien und ein weiterer aus China sind betroffen. Entsprechende Fertigarzneimittel wurden weltweit zurückgerufen.
Inzwischen wurden verschiedene Risiken berechnet und es wurde nach Verantwortlichen gesucht – so richtig sicher weiß man aber noch nichts. Und während man noch nichts Genaues weiß, gesellen sich immer neue Fragen zum Fall Valsartan hinzu: So kann man noch nicht ausschließen, dass neben Valsartan weitere Sartane von einer Nitrosamin-Verunreinigung betroffen sind. Am 30. August kündigte die US-amerikanische Arzneimittelbehörde FDA an, auch alle anderen Sartane auf ihre Syntheseschritte und die Gefahr der NDMA-Bildung hin zu untersuchen. Die EMA wurde vergangenen Freitag in Losartan fündig. Hingegen war es wieder die FDA, die schon zuvor zwei 483-Formulare ins Internet gestellt hatte, in denen zwei Inspektionen durch die US-Behörde aus den Jahren 2016 und 2017 bei Zhejiang Huahai dokumentiert wurden.
Mehr zum Thema

Zhejiang Huahai Pharmaceutical
Die FDA fand schon 2017 eine unbekannte Verunreinigung beim Valsartan-Hersteller
Um welche Wirkstoffe es bei der FDA-Inspektion 2017 ging, ist nicht bekannt. Dennoch erinnern die im Dokument aus dem Jahr 2017 beschriebenen Mängel an den Fall Valsartan: Von nicht erklärbaren und unbekannten Verunreinigungen war dort die Rede, die als Ausschläge im Chromatogramm bei der Qualitätskontrolle auffielen. Zhejiang Huahai nannte sie „ghost peaks“. Und Europa? Waren neben der FDA auch europäische Behörden vor Ort in China? Ja, sogar eine deutsche Behörde und diese hat DAZ.online um Stellungnahme gebeten.
Sartane waren Gegenstand einer deutschen Inspektion
In der europäischen EudraGMDP-Datenbank sieht man, wann welche Behörde die Einhaltung der GMP-Richtlinien bei welchem Hersteller überprüft hat – und mit welchem Ergebnis. Aus der Datenbank geht hervor, dass eine deutsche Inspektion zuletzt am 6. April 2017 bei Zhejiang Huahai in China stattgefunden hat. Die Inspektion erfolgte durch die Hamburger Behörde für Gesundheit und Verbraucherschutz, die Zhejiang Huahai in den vergangenen Jahren mehrfach inspiziert hat. Um Sartane ging es im April 2017 jedoch nicht.
Ein weiterer Eintrag in der Inspektionsdatenbank bezieht sich aber auf eine Inspektion der Hamburger Aufsicht aus dem Jahr 2015, damals ging es auch um Sartane bei Zhejiang Huahai. Allerdings nicht um Valsartan, sondern um Candesartan, Irbesartan und Telmisartan. Die Inspektion dauerte vier Tage an und erfolgte durch zwei GMP-Inspektoren nach den Vorgaben des Good Manufacturing Practice Leitfadens der EU.
2012 haben die Hamburger auch die Valsartan-Produktion überprüft
Fand also nie eine Inspektion der Valsartan-Produktion bei Zhejiang Huahai durch eine deutsche Behörde statt? Doch. Denn in der Datenbank gibt es eine weitere Inspektion mit dem Enddatum vom 27. September 2012. Die damals inspizierten Arzneimittel waren laut dem Dokument: Candesartan, Donepezil, Irbesartan, Telmisartan, Voriconazol und auch Valsartan. Größere Mängel scheint es bei dieser Inspektion nicht gegeben zu haben. Denn: Die Hamburger Behördenmitarbeiter stellten ein GMP-Zertifikat aus. Das ausgestellte Dokument galt bis zum 27. September 2015. Ob die Behörde damals eventuell kleinere Mängel festgestellt hat und wie diese behoben wurden, blieb gegenüber DAZ.online bislang unbeantwortet.
Fest steht aber: Zumindest einmal gesehen hat also auch die Hamburger Behörde die Valsartan-Herstellung. Was dabei auffällt, ist das Datum: das Jahr 2012. Denn ausgerechnet im Jahr 2012 sollen die Änderungen im Syntheseweg eingeführt worden sein, von denen angenommen wird, dass sie zur Bildung von NDMA als Nebenprodukt geführt haben.
Hätte die Hamburger Behörde Verunreinigungen finden können?
Hätten also verdächtige Ausschläge im Chromatogramm wie bei
der FDA 2017 auch 2012 schon bei einer GMP-Inspektion durch die deutsche Behörde auffallen können? Auf die generelle Frage nach Verunreinigungen antwortet die Hamburger Behörde so:
Der Syntheseweg und damit die zu spezifizierenden Verunreinigungen sind bereits durch die zuständigen Zulassungsbehörden genehmigt. Eine erneute inhaltliche Prüfung soll und kann im Rahmen einer GMP-Inspektion nicht stattfinden."
Sprecher der Hamburger Behörde für Gesundheit und Verbraucherschutz
Die Frage, ob die durch die Zulassung festgeschriebenen Qualitätskontrolluntersuchungen geeignet sind, stelle sich im Rahmen einer GMP-Inspektion somit „explizit“ nicht.
Auch wenn der Syntheseweg im Rahmen einer GMP-Inspektion nicht geprüft werden kann und soll, heißt das dann, dass bei einer solchen Inspektion auch keine unerwarteten Verunreinigungen auffallen können? Auch diese Frage blieb bislang unbeantwortet.
Hat die FDA besser gesucht als die Europäer?
Die FDA hat 2017 im Rahmen von GMP-Inspektionen jedenfalls bei einem unbekannten Wirkstoff Verunreinigungen gefunden. Wenn auch europäische Behörden bei dem chinesischen Hersteller genauer hingeschaut haben, stellt sich also die Frage, warum nur die FDA fündig geworden ist. Die EMA kommentierte das gegenüber DAZ.online so: Die FDA-Inspektion sei der EMA bekannt, doch habe es zu diesem Zeitpunkt keine Sicherheitsbedenken gegeben. Aufmerksam auf die FDA-Inspektionen bei Zhejiang Huahai in den Jahren 2016 und 2017 sei die EMA aber erst während der Prüfung des Valsartan-Falls geworden. Es wird weiterhin darauf hingewiesen, dass in einem 483-Formular der FDA nur Beobachtungen festgehalten werden, die nicht automatisch bedeuten, dass der Produktionsstandort nicht GMP-konform arbeitet.
Sind europäischen Behörden bei Zhejiang Huahai nie Mängel aufgefallen?
Die letzte europäische Inspektion bei Zhejiang Huahai fand im März 2018 statt – durch die italienische Arzneimittelbehörde. Gegenstand dieser Inspektion waren jedoch keine Sartane, sondern die Wirkstoffe Enalapril, Rivaroxaban und Quinapril. Schwerwiegende Mängel sind auf Basis der EudraGMDP-Datenbank im Fall von Zhejiang Huahai auch im Rahmen dieser Inspektion nicht erkennbar. Und auch insgesamt werden in der Datenbank keine „Non-Compliance-Reports“ für Zhejiang Huahai gelistet. Das heißt allerdings nicht, dass es bei Zhejiang Huahai von europäischer Seite in den vergangenen Jahren nie etwas zu kritisieren gab. Denn die Hamburger Behörde erklärt:
Dass in der Liste keine Mängel aufgeführt sind, kann auch bedeuten, dass entdeckte Mängel bereits im Rahmen eines festgelegten Verfahrens durch die Firma unter Vorlage von Nachweisen beseitigt wurden."
Sprecher der Hamburger Behörde für Gesundheit und Verbraucherschutz
Warum reiste ausgerechnet die Hamburger Behörde regelmäßig nach China?
Aber wie kam es überhaupt dazu, dass gerade die
Hamburger Behörde für Gesundheit und Verbraucherschutz Zhejiang Huahai
regelmäßig inspizierte? Das erklärt die Behörde gegenüber DAZ.online so: „GMP-Inspektionen in einem sogenannten Drittland
werden aufgrund von Anträgen durchgeführt. In diesen Anträgen sind die zu
inspizierenden Arzneimittel oder Wirkstoffe zu benennen.“ Der
Eudra-GMDP-Datenbank zufolge erfolgten diese Anträge durch die Firma Alfred E. Tiefenbacher (AET) aus Hamburg. Diese hat sich eigenen Angaben zufolge die Beschaffung und den europaweiten Vertrieb von APIs (Active Pharmaceutical Ingredient), also Wirkstoffen, auf die Fahne geschrieben.
Mehr zum Thema

Zur Bedeutung des Written-Confirmation-Verfahrens im aktuellen Valsartan-Fall
Überwachung der Länder ist außen vor
Valsartan-Krise
BMG: Behörden dürfen Pharmawerke in China inspizieren



0 Kommentare
Das Kommentieren ist aktuell nicht möglich.